Hi MRtrixers.
I am trying the new mtnormalise command on a single subject but am faced with an error:
mtnormalise fod_wm.mif fodwm_norm.mif gm.mif gm_norm.mif csf.mif csf_norm.mif -mask mask.mif
mtnormalise: [done] loading input images
mtnormalise: [ERROR] Non-positive tissue balance factor was computed. Tissue index: 2 Balance factor: 0 Needs to be strictly positive!
Can you please advise me as to what the error represents?
Thankyou.
Jerome
Hi @JMaller1,
That shouldn’t typically happen for a normal scenario; so there must be something special about yours. For the normalisation process, the algorithm tries to rebalance the tissues so they represent similar magnitudes somehow. It essentially tries to use them a bit more like actual fractions (even though they still aren’t). In your case, the CSF one (that’s tissue index 2 there) found itself with a balancing factor of 0, which is quite strange. If it would go negative, it’d typically end up with an actual negative number… for it to hit exactly 0… well, I’m not sure what caused that. Let’s start first by investigating when exactly it happened during the algorithm. Can you run the exact same command again, yet add the -info option to it, and provide us with the complete command line output that follows?
Cheers,
Thijs
Just adding to that immediately, after a brief discussion with @rtabbara as to what may have been the actual cause here: a possible situation leading to exactly this outcome could be that your CSF image is all zero within the supplied mask. So it could be worthwhile checking:
- if the
csf.mif image is indeed all zero
- if your
mask.mif image is a while brain mask, and for instance not just a small region (which may not include CSF, so csf.mif is then indeed all zero within the mask)
So as advise, if this would be the case: make sure mask.mif is a whole-brain mask, and make sure all tissue types are in fact present.
Thankyou so much for your very prompt response.
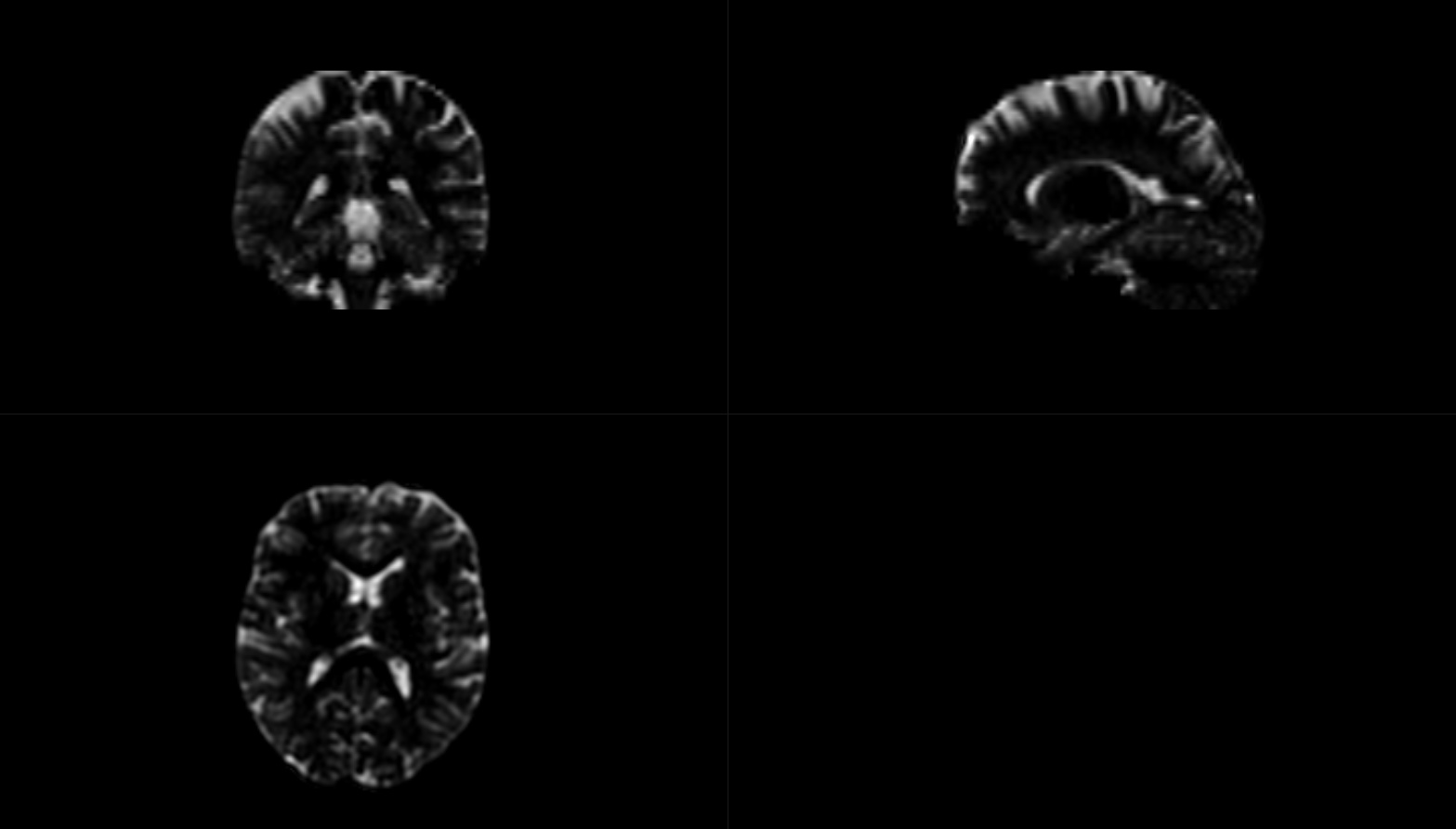
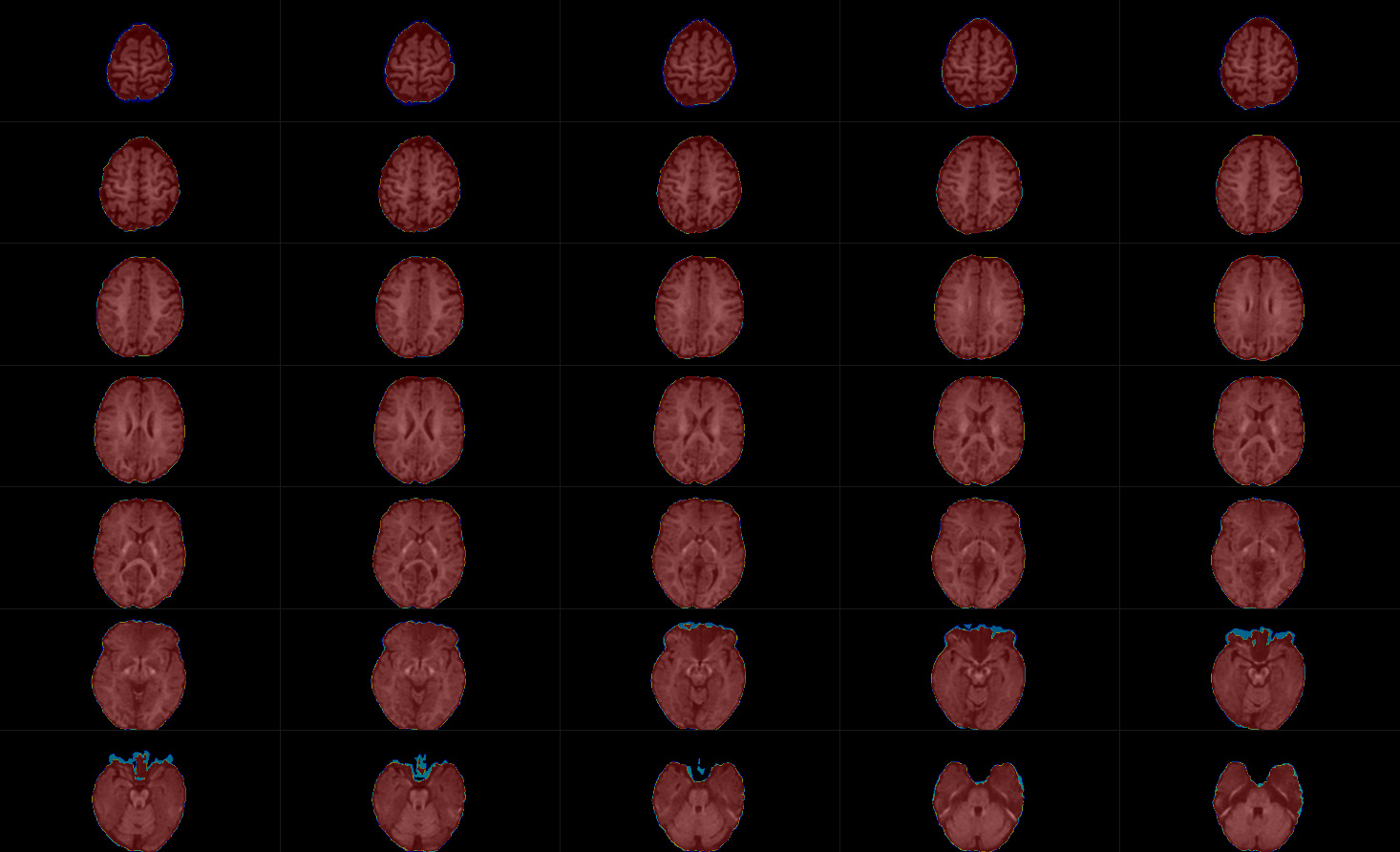
The mask seems to covers the entire brain, and the CSF file is non-zero (see figures below).
I have now repeated the command but added -info at the end:
$ mtnormalise fod_wm.mif fodwm_norm.mif gm.mif gm_norm.mif csf.mif csf_norm.mif -mask mask.mif -info
mtnormalise: [. ] loading input images…
mtnormalise: [INFO] opening image “fod_wm.mif”…
mtnormalise: [INFO] image “fod_wm.mif” opened with dimensions 256x256x48x45, voxel spacing 1.0156000000000001x1.0156000000000001x2.5000399999999998x3.9100000000000001, datatype Float32LE
mtnormalise: [INFO] opening image “gm.mif”…
mtnormalise: [INFO] image “gm.mif” opened with dimensions 256x256x48x1, voxel spacing 1.0156000000000001x1.0156000000000001x2.5000399999999998x3.9100000000000001, datatype Float32LE
mtnormalise: [INFO] opening image “csf.mif”…
mtnormalise: [INFO] image “csf.mif” opened with dimensions 256x256x48x1, voxel spacing 1.0156000000000001x1.0156000000000001x2.5000399999999998x3.9100000000000001, datatype Float32LE
mtnormalise: [INFO] opening image “mask.mif”…
mtnormalise: [INFO] image “mask.mif” opened with dimensions 256x256x48, voxel spacing 1.0156000000000001x1.0156000000000001x2.5000399999999998, datatype Int16LE
mtnormalise: [done] loading input images
mtnormalise: [INFO] Iteration: 1
mtnormalise: [ERROR] Non-positive tissue balance factor was computed. Tissue index: 2 Balance factor: 0 Needs to be strictly positive!
I tried the mtnormalize command on another subject’s data and it worked well, so I’m not sure why this subject’s dataset produces errors…
Regards,
Jerome
Thanks for the detailed report @JMaller1. The images shown do look completely normal indeed. I was about to ask you to share the data so we could have a look at it, until I did a quick test myself over here. I figured out that I’d made a mistake suggesting the CSF would be the problem here (I reasoned the tissue index started counting from 0, but I think I had originally changed that in the code myself to start counting from one). Essentially, tissue index 2 relates to your second input tissue type, which in your case is GM. That, together with the CSF and WM images you posted, makes me suspect the GM is actually going to be zero (or very close to it). I’m quite used to looking at multi-tissue reconstructions these days, and your CSF looks like it is a tiny bit brighter in GM regions that I would expect it to be; but more importantly, your WM looks like it gets a lot of GM. What I suspect is a de facto 2-tissue reconstruction… which may be a realistic guess if you’ve potentially only used single-shell data to do multi-tissue CSD.
So, long story short: can you also check your GM input image? I predict it’s going to be all zero or amazingly small magnitudes (both positive as well as slightly negative potentially).
If that is the case, and you only have single-shell data, then at the moment (until I release SS3T-CSD at some point) you’re limited to 2-tissue WM and CSF CSD. There’s other good news there for you though: I did start to test extensively whether mtnormalise still works and makes sense on 2-tissue WM and CSF reconstructions, and the answer so far is “yes”. So essentially, you can just (consistently) provide only the WM and CSF to mtnormalise, and it would make sense. Also, you don’t have to ask dwi2fod msmt_csd for the GM in this scenario, since it won’t give you any by definition. Those subjects where it did succeed to run, may have had quite weird or problematic outputs as well with a GM compartment that is numerically very close to zero.
If you want more information on such a 2-tissue strategy, I recommend to read this post and all the replies, as well as all the links to other posts I have given in the replies I made to that post: Fixel-Based Analysis on single shell data
Hope this helps!
Cheers,
Thijs
1 Like
Thankyou so much Thijs! I tried the mtnormalize without the gm and it worked beautifully. I am used to working with only multishell data, so this single shell data was a bit of a challenge to me with mtnormalize.
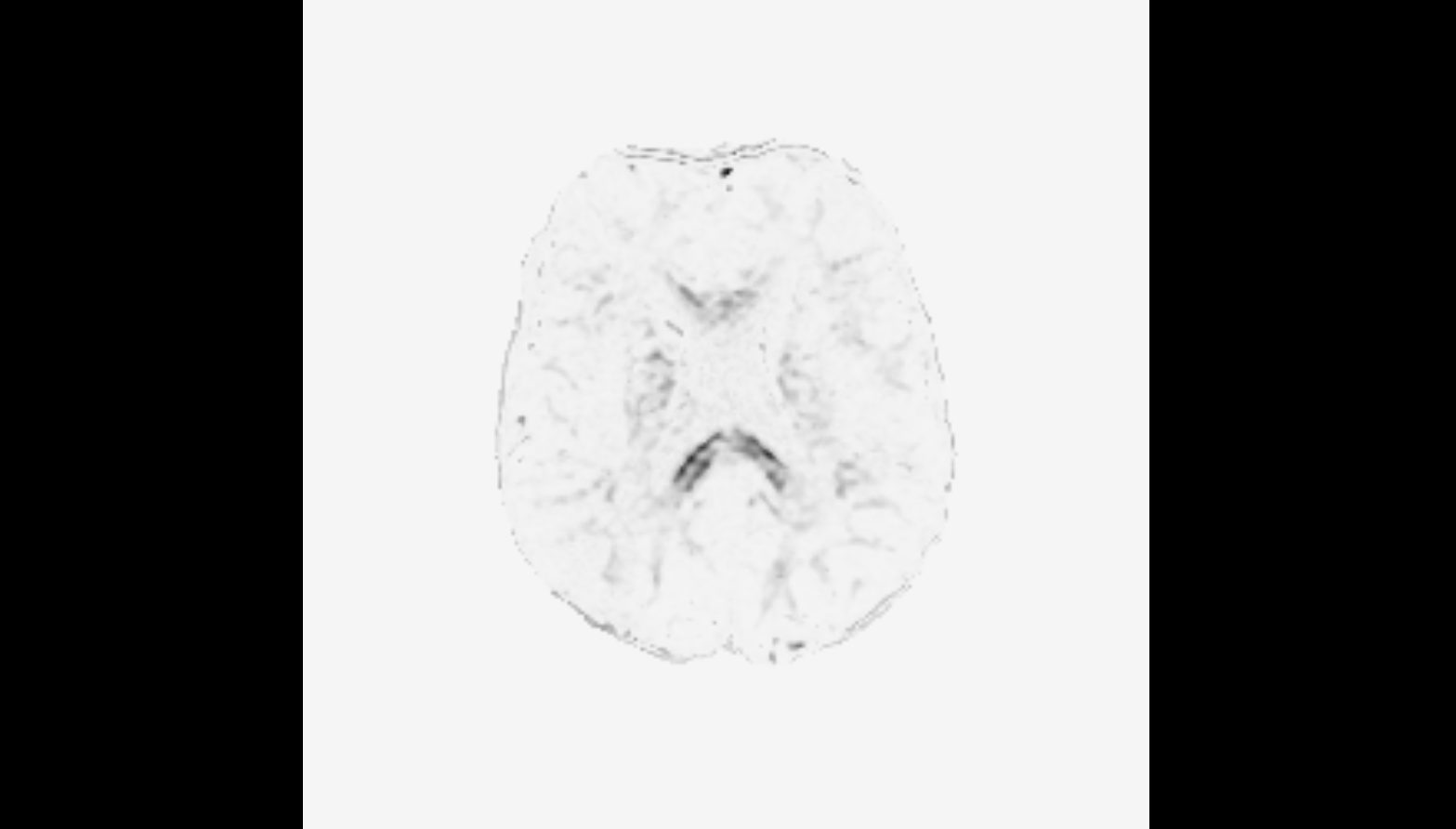
FYI, here is a pic of the gm.mif. Indeed, most is very close to zero.
Thanks again.
Jerome
1 Like