Hi MRtrix community,
Our aim is to do msmt csd but before I dive in to my question I will explain what data I have:
we scanned some entire heads on a 3T siemens scanner, for days at a time. We have b values of 4000 (ap only) and 8000 (ap and pa) along with a bunch of B0s per head. The same gradient tables where used for all sets of bvalues and added to the image headers properly. and the echo time was adjusted to compensate for signal changes occurring due to length of fixation per head (as heads were not fresh and differed in fixation time).
and now the preprocessing i did:
- preliminary DTIFIT to check directions per bvalue - was fine
- denoise and degibbs via mrtrix with combined bvalues- also fine
- run topup on combined ap pa b0s manually via fsl - fine
- no applytopup - straight to eddy to correct the entire combined dataset - alright
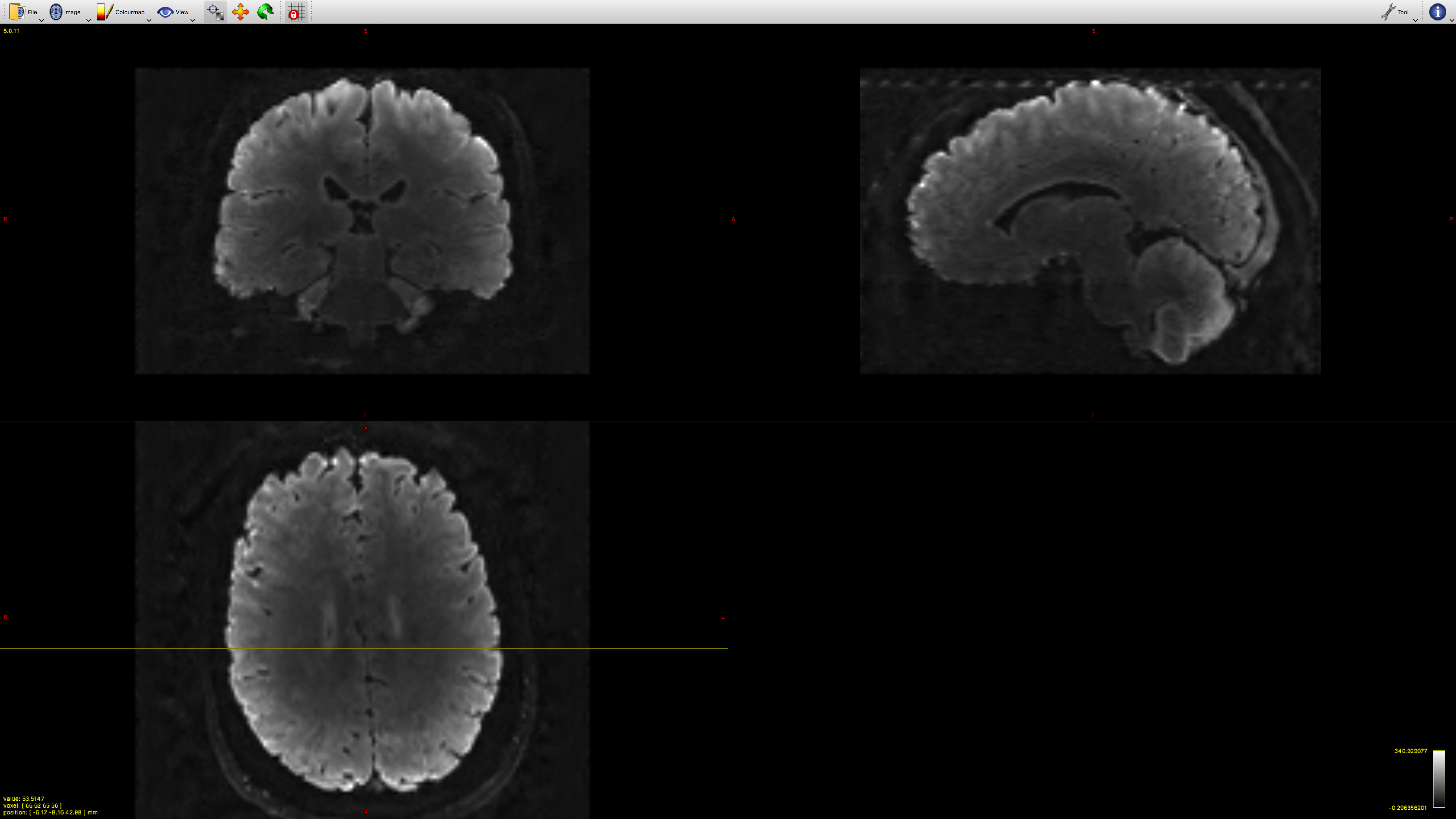
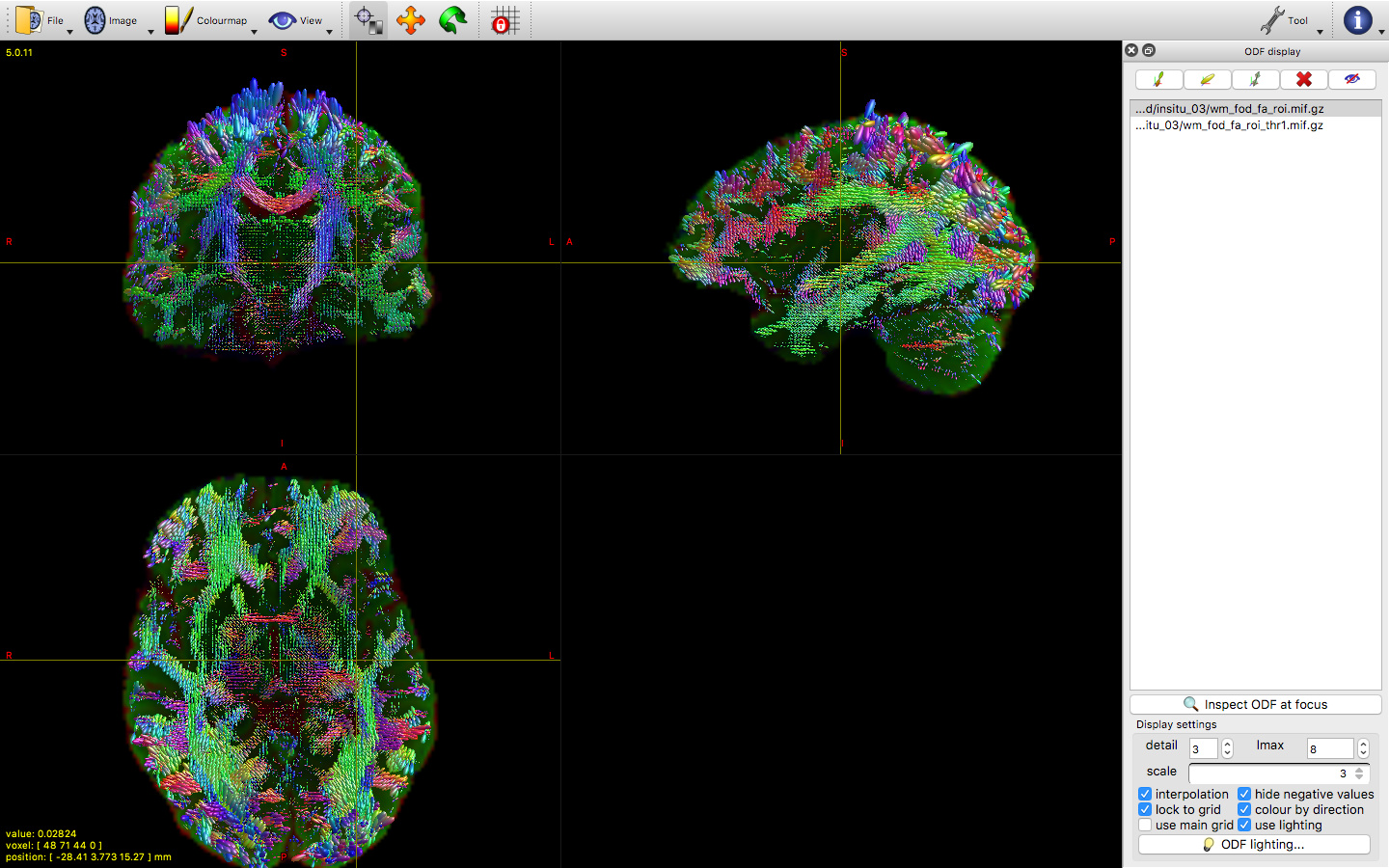
However when we run RFEs and generate the subsequent FODs we have some artefacts specifically along the cortex (this could make sense as its ‘more fixed’ than deeper tissues). But im not sure how to deal with it. The level of noise is obviously not constant across bvalues.
we initially ran dhollander to segment the tissue but later decided to do it manually by selecting voxels for GM and basing the WM rois on the FA map (CSF is a bit arbitrary because its more background as its mostly just water) and the tissue segmentation is quite good. but the FOD values for WM become huge along the motor-ish and occipital cortical areas. It also happens regardless of how strict/liberal we are with the fa values.
This is accompanied by a bright artefact along the tissue. I am reluctant to bin volumes where the artifact is at its worst because its relatively constant across the scan. Also we could i guess just mask that area out but I’m not keen on that either. N4biascorrections via ANTs wont work as it outputs an average image.
all tips, tricks and questions are gratefully received
cheers!
Beth