Hi all,
I am using the following command:
dwi2response dhollander ${WP}/eddy_corrected_dataup.nii.gz -fslgrad ${WP}/eddy_corrected_dataup.eddy_rotated_bvecs ${WP}/dataup.bval -lmax 6,6 -mask ${WP}/hifi_nodifupdn_brain_mask.nii.gz -shell 0,700 ${WP}/response_wm.txt ${WP}/response_gm.txt ${WP}/response_csf.txt
eddy_corrected_dataup.nii.gz has 6 b0 volumes at the beginning followed by 61 diffusion volumes.
The b-value is 700.
The content of the response files are:
response_csf:
2086.740266636972
95.50751661620647
I’m not 100% sure what’s causing that issue, though I have a strong suspicion. Would it be possible for you to provide me with some example data so I can reproduce the issue?
The -nan values are wrong indeed (and that output should certainly not be used). The problem is simply due to the -lmax 6,6 option you specified. Nowadays, for data like yours, you don’t even have to specify the -lmax option at all; by default it’ll estimate the responses up to lmax=10 for you, which works perfectly even for data with “apparently” too few directions. Don’t worry about it. Just removing the -lmax 6,6 option will solve your problem. If you want, you can always report back here with the output you get when removing the -lmax 6,6 option, and I’ll be able to reassure you whether the output makes sense.
Cheers,
Thijs
(Side-note: If for some very specific reason (but again, generally, this is not needed at all) you’d want to limit the WM response to lmax=6, the correct option to specify would be -lmax 0,6 in your case. The b=0 data can/should only be estimated isotropically, hence the first “0” in that option.)
The response_wm.txt contains five zeros, like this:
cat response_wm.txt
1941.282167282583 0 0 0 0 0
1018.477590045386 -290.8834011909341 37.78147727948814 -3.397613063674172 -0.6472425603888992 0.7481814258406785
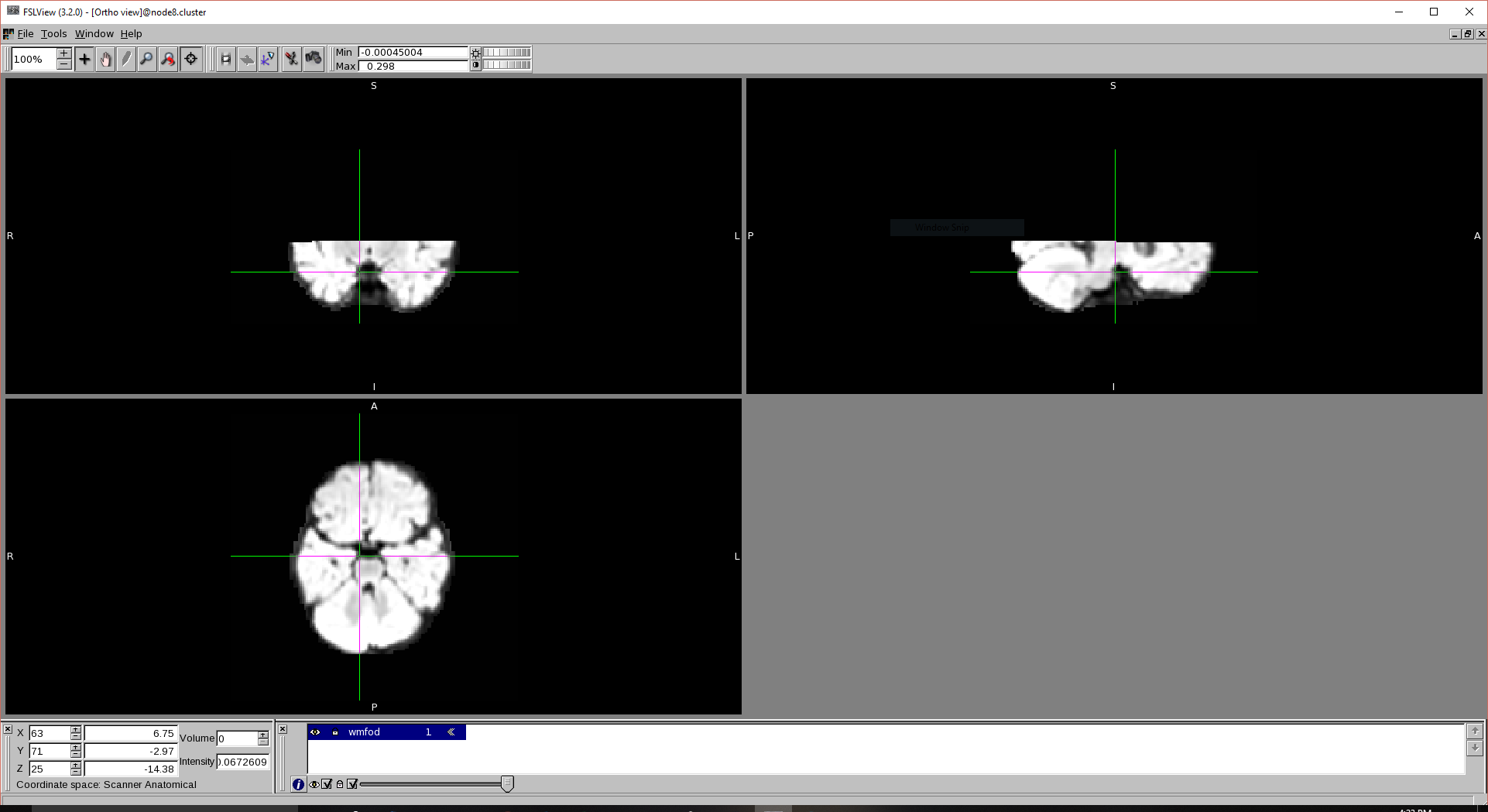
and the wmfod image is chopped in half like below:
No worries about those zeros, that’s entirely expected (and should even be like that). See also @jdtournier’s reply to your other post here: Contents of response_*.txt , which may help you to understand these zeros. The first line of your response_wm.txt is essentially the b=0 part of the WM response function. We don’t expect ( / want) any anisotropy at b=0, so only the lmax=0 term is non-zero. The non-zero lmax=0 term encodes the overall size of that shell of the response, whereas the lmax=2,4,6,8,10 terms encode the shape. If the latter are all zero, you’ve got an isotropic shell, which is exactly what the b=0 shell is.
That’s entirely separate to your second issue, the chopped WM FOD image. I don’t know for sure what happened there, but it appears either your dwi2fod step crashed or otherwise stopped prematurely, or the result has not been written to disk yet. The latter may be the case if you work with a remote storage, under certain circumstances. Did you get an error / crash from dwi2fod? Are you sure it wasn’t still running at the time you opened that image in mrview? If it all finished successfully, and you subsequently open the WM FOD file (again), you shouldn’t get this. (the only other explanation would be that your brain mask stops right there, but that’d be weird in its own right)
The dwi2fod does not output any error and it is done when I open the wmfod.
My first thought was that there might be a problem with mask but I checked it and it looks good.
One thing I should mention is that because I’m running mrtrix on our cluster and I can’t run mrview over the ssh
I convert the .mif to .nii.gz whenever I want to check the images so I can review them in FSL.
How you would normally debug the dwi2fod? what and where I should look for logs or intermediate files?
Well, definitely have a look at the intermediate files, so the problem can be narrowed down to a specific step. I struggle to see how the conversion to .nii.gz could cause issues. Maybe the FSL viewer itself is the problem, but again I’m not sure how that could be explained… So first things first: try and get the .mif output of the (successfully completed) dwi2fod on a computer / system that has mrview, and take a look at the .mif directly in mrview.
Nope, doesn’t help. But it’s also unrelated: this is output from the subsequent mtnormalise step. There’s a few explanations for that error, but it’s best to resolve the original problem first. If something is indeed wrong with the .mif output above (but mrview can confirm that or rule it out), then it’s not worth to try and resolve this mtnormalise error at this stage. If the .mif files of all tissue types do look fine however, and you’re still getting this mtnormalise error, we should be looking into that separately.
Already getting ahead of myself anyway though about the separate mtnormalise problem / error: I recall from other posts you’re working on single-shell data (i.e. single non-bzero b-value, and b=0 image(s)), right? Running dwi2response dhollander on that is 100% fine, and in case of single-shell + b=0 data gives you 3 response functions (WM, GM, CSF) for that. Nothing wrong at that stage. But (big but here), running dwi2fod msmt_csd, with 3 tissue types, yet only with single-shell + b=0 data, is not enough. With “not enough”, I mean that you’ll still be able to run it, and get an output, but typically the GM volume will be all zero (apart from small numerical deviations). Putting that near-zero GM volume together with the WM and CSF volumes into mtnormalise though, becomes the problem, and (without going into the technical details why) typically results in that error. Also, even if it wouldn’t result in that error, the output of such an operation is inappropriate. Long story short here: with single-shell + b=0 data, at the moment (without SS3T-CSD being available), you should only perform 2-tissue CSD, for which you can still use e.g. the WM and CSF response functions from dwi2response dhollander. See also the post I linked to in my other reply (that has the long explanations on the response function algo’s and CSD algo’s); and further links in that linked post.
But well, this long paragraph getting ahead of myself is really only relevant at all if the original problem is either solved (or was just a visualisation issue in FSL, or mistake in the conversion to .nii.gz).