Hi,
I have been trying to use the script dwi2response but I get an error message and I don’t know why:
I did:
dwi2response msmt_5tt DWI.mif 5tt.mif wm.txt gm.txt csf.txt
(after obtaining the files needed: specific tissue type segmentations, compiled in 5tt.mif, and DWI file in .mif format, containing bvec information)
And I got:
dwi2response:
dwi2response: Note that this script makes use of commands / algorithms that have relevant articles for citation; INCLUDING FROM EXTERNAL SOFTWARE PACKAGES. Please consult the help page (-help option) for more information.
dwi2response:
dwi2response: Generated temporary directory: /tmp/dwi2response-tmp-8EEIIY/
Command: mrconvert 5tt.mif /tmp/dwi2response-tmp-8EEIIY/5tt.mif -quiet
Command: mrconvert DWI.mif /tmp/dwi2response-tmp-8EEIIY/dwi.mif -stride 0,0,0,1 -quiet
dwi2response: Changing to temporary directory (/tmp/dwi2response-tmp-8EEIIY/)
Command: dwi2mask dwi.mif mask.mif -quiet
Command: dwi2tensor dwi.mif - -mask mask.mif -quiet | tensor2metric - -fa fa.mif -vector vector.mif -quiet
Command: mrtransform 5tt.mif 5tt_regrid.mif -template fa.mif -interp linear -quiet
Command: mrconvert 5tt_regrid.mif - -coord 3 0 -axes 0,1,2 -quiet | mrcalc - 0.95 -gt fa.mif 0.2 -lt -mult mask.mif -mult gm_mask.mif -quiet
Command: mrconvert 5tt_regrid.mif - -coord 3 2 -axes 0,1,2 -quiet | mrcalc - 0.95 -gt mask.mif -mult wm_mask.mif -quiet
Command: mrconvert 5tt_regrid.mif - -coord 3 3 -axes 0,1,2 -quiet | mrcalc - 0.95 -gt fa.mif 0.2 -lt -mult mask.mif -mult csf_mask.mif -quiet
dwi2response: Calling dwi2response recursively to select WM single-fibre voxels using ‘tournier’ algorithm
Command: dwi2response -quiet tournier dwi.mif wm_ss_response.txt -mask wm_mask.mif -voxels wm_sf_mask.mif
fixel2voxel: [ERROR] fixel image is empty
dwi2response: [ERROR] Command failed: fixel2voxel iter0_peaks.msf split_value iter0_amps.mif -quiet
dwi2response: [ERROR] Command failed: dwi2response -quiet tournier dwi.mif wm_ss_response.txt -mask wm_mask.mif -voxels wm_sf_mask.mif
dwi2response: Changing back to original directory (/mounts/auto/carmen_connectivity/scon_BL_Sep2016/subjects/complete_dataset/mytest)
dwi2response: Deleting temporary directory /tmp/dwi2response-tmp-8EEIIY/
It seems to complain about the fixel image, but in the website I cannot see any requirement in this sense.
Any help will be welcome.
Thank you very much in advance.
Best,
Carmen
Hi @carmentur,
Welcome to the MRtrix community forum, and thanks for your interest in using MSMT-CSD. The problem you’re experiencing is similar to the one described in this topic, which was a problem with the 5tt file in that case. Alternatively, it may be that your b-values come in different units; which may confuse the automatic shell detection. To check on the latter, could you provide us with the output of this command:
mrinfo DWI.mif -shells -shellcounts
To check the former, you can actually avoid using a 5tt image at all, using the brand new “dhollander” algorithm in dwi2response. Simply make sure your MRtrix installation is reasonably up to date (the algorithm was added only about 3 weeks ago), and try this one:
dwi2response dhollander DWI.mif wm.txt gm.txt csf.txt
Note that the latter algorithm not only involves less hassle (i.e. is not dependent on a T1, subvoxel accuracy coregistration with the diffusion MRI data, and 5tt segmentation), but was actually found to be more accurate as well, probably (partially) due to not being dependent on the aforementioned processing steps. For more information, see this very recent abstract on the method: https://www.researchgate.net/publication/307863133_Unsupervised_3-tissue_response_function_estimation_from_single-shell_or_multi-shell_diffusion_MR_data_without_a_co-registered_T1_image
If you still run into the same error, the problem will probably be with the shell selection, in which case the mrinfo -shells -shellcounts output should give us the information to figure that out.
Cheers,
Thijs
Hi Thijs,
Thank you very much for the reply.
I have tried the command you suggested: mrinfo DWI.mif -shells -shellcounts
And I got:
0 300.027 711.002 1999.7
7 8 15 30
which is correct.
I tried the other command: dwi2response dhollander DWI.mif wm.txt gm.txt csf.txt
But I am afraid my MRtrix version doesn’t have this new function ‘dhollander’.
I will make sure I have the latest version of the software installed and I will get back to you if I encounter any problems.
Thanks again!
Best wishes,
Carmen
Dear Thijs,
The problem has now been resolved: dwi2response dhollander works perfectly!
Thanks so much.
Best,
C
Hi @carmentur,
Thanks for those pieces of information; it does give us insight into what was probably the original problem: I’d say it’s very likely something that had to do with the 5tt file.
As you noticed already as well, those b-values and the numbers that go with them definitely seem correct and in the appropriate units; so no worries there at all. That’s basically all the new “dhollander” algorithm needs. Given data of a fully developed (i.e. 3 years of age or more, more or less) human brain, the algorithm should do at least as well as the “msmt_5tt” algorithm, and in quite a few cases even better (see the aforementioned abstract). Just to say, you can happily stick with it, if the output leads to good MSMT-CSD results further down the pipeline.
We’re always happy to give those results a check as well; if you want to, feel free to post a screen shot of the WM, GM and CSF outcomes after MSMT-CSD!  To be honest, I’m actually (personally) quite curious to see the quality of the result given that particular acquisition (i.e. b-values and numbers of directions); it seems to be a slightly unusual setup (as in: I’m not used to seeing it myself). Any particular reason(s) why/how those b-values and numbers of directions were decided upon?
To be honest, I’m actually (personally) quite curious to see the quality of the result given that particular acquisition (i.e. b-values and numbers of directions); it seems to be a slightly unusual setup (as in: I’m not used to seeing it myself). Any particular reason(s) why/how those b-values and numbers of directions were decided upon?
Cheers,
Thijs
Hi Thijs,
Thank you for your reply.
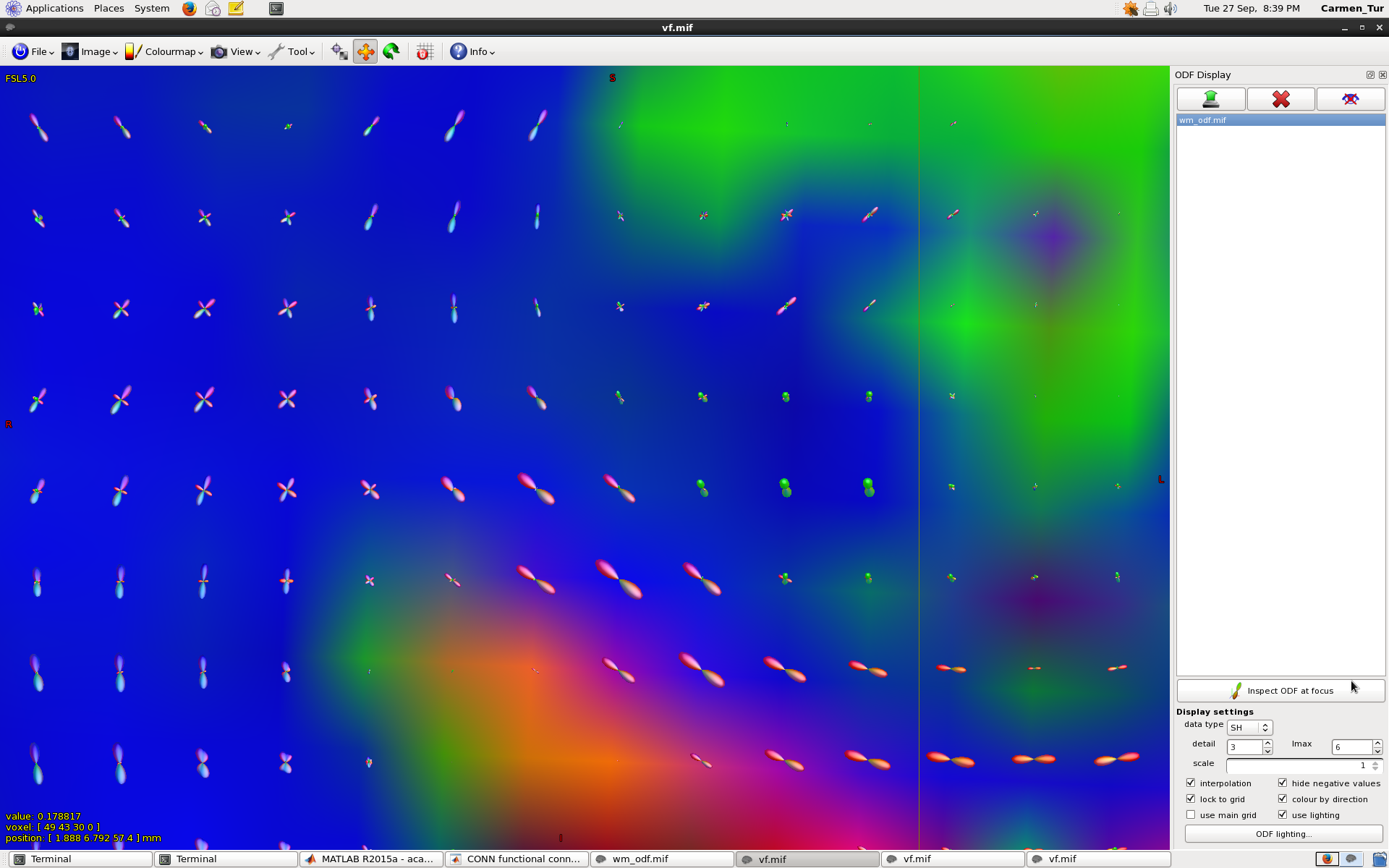
Please see attached 1 screenshot where you can see the corpus callosum and corona radiata 
The DWI protocol was decided a while ago, before my start in my current research group. It was not initially designed to do tractography, but it may still be used for that!
Thanks again
Best,
Carmen
Hi again Carmen,
Those FODs definitely look ok, given that acquisition setup! By the way, if you want to enlarge them a bit for visualisation purposes, you can always use the “scale” box in the “ODF display” tool (somewhere at the bottom right of the screen shot you posted).
Maybe a final check to ask (I’m quite curious to see how well my new response function selection algorithm worked for you): would you mind posting the response functions you obtained? So just a simple copy-paste of the contents of the wm.txt, gm.txt and csf.txt files.
But again, it definitely looks like everything went very well. 
Cheers,
Thijs
Hi Thijs,
Thanks again for your message and interest!
This is the content of the text files with the response function:
wm:
353366.5151 0 0 0
272642.2966 -30477.95461 0 0
206019.9626 -52933.83031 7490.906059 0
117606.3785 -64342.3779 20906.04635 -4836.539719
gm:
493897.810463
359361.473363
254958.978674
98694.8387984
csf:
1430618.40122
418327.468075
121288.662527
6419.26066275
Thanks again!
Carmen
Just double-checked those, and they look ok! Thanks again for reporting back, this provides me with a few useful insights.
Cheers,
Thijs
 To be honest, I’m actually (personally) quite curious to see the quality of the result given that particular acquisition (i.e. b-values and numbers of directions); it seems to be a slightly unusual setup (as in: I’m not used to seeing it myself). Any particular reason(s) why/how those b-values and numbers of directions were decided upon?
To be honest, I’m actually (personally) quite curious to see the quality of the result given that particular acquisition (i.e. b-values and numbers of directions); it seems to be a slightly unusual setup (as in: I’m not used to seeing it myself). Any particular reason(s) why/how those b-values and numbers of directions were decided upon?