Dear experts,
we are currently setting-up a new DWI protocol for our new Siemens Vida 3T scanner.
- Purposes: CSD-based tractography, structural connectivity, FBA
- Aim: best possible angular and spatial resolution
- DWI measurement time plan: approx 15 minutes
- Multiband at the disposal.
Based on discussions on this forum and recommendations in publications, especially
we tried to set following DWI scheme on our product DWI sequence using custom diffusion directions:
gen_scheme 2 0 12 1000 40 3000 160
i.e. diffusion scheme split to 2 separate series with opposite phase encoding direction (AP/PA).
The aim was to dedicate most possible time to gain SNR on b=3000. b=1000 (instead of something around b=1500) was selected due to retain ability to fit diffusion tensor model on the data, if needed.
Main measurement parameters:
MB factor 3, voxel size 2x2x2 mm3, 69 slices, TR=4500 ms, acquisition duration 2x8.7 minutes.
To assess the quality of the data and SNR requirements using selected voxel size, I tried to evaluate SNR of b=0 and b=3000 shell.
I read recommendations not to go below SNR = 15 in b=0 shell by @jdtournier
and aim for SNR>5 in most outer shell by @dchristiaens (althought, he, admits, it is difficult to achieve).
I am using following commands to get SNR, partially adapted from this thread:
dwidenoise dwi.mif dwi_denoised_extent_7.mif -extent 7x7x7 -noise noise_extent_7.mif
dwi2mask dwi_denoised.mif mask.mif
Concerning noise estimation, I found two recommendations:
- using voxel-wise std of b=0
dwiextract -shell 0 dwi_concat_denoised_extent_7.mif - | mrmath - std -axis 3 - | mrstats -
volume mean median stdev min max count
[ 0 ] 15.1416 5.64851093 27.0527 0 785.402 690000
- using estimated std of noise from
dwidenoise
mrstats -output mean -mask mask.mif noise_extent_7.mif
10.6097
The results are quite different…
Signal in shells:
dwiextract -shell 0 dwi_concat_denoised_extent_7.mif - | mrstats -output mean -mask mask.mif -allvolumes -
223.168
dwiextract -shell 1000 dwi_concat_denoised_extent_7.mif - | mrstats -output mean -mask mask.mif -allvolumes -
78.423
dwiextract -shell 3000 dwi_concat_denoised_extent_7.mif - | mrstats -output mean -mask mask.mif -allvolumes -
33.7226
Thus, SNR, using method 1 in shells is
b=0: 14.7
b=1000: 5.2
b=3000: 2.22
usind method 2
b=0: 21
b=1000: 7.4
b=3000: 3.1
Could you please comment on on our prepared protocol?
-
Is the SNR, in your opinion, sufficient? SNR>5 condition in outer shell is not fullfilled. Should we rather increase voxel size to get better trade-off between SNR and angular/spatial resolution?
-
Is the rather large amount of directions for b=3000 beneficial to gain SNR, regarding possible pitfall with rician bias on low SNR, as mentioned in following thread?
SNR calculation comparable to dipy? - #5 by jdtournier
Or should we rather spend acquisition time by acquiring additional b shell? I was considering to add the shell to be able to model additional compartment as mentioned here:
Good b-value sensitivity parameter for 2shell - #2 by jdtournier -
What method, from the listed two, do you recommend for noise estimation, seeing the fact that each produces different result?
Further recommendation was on optical inspection of fibre responses and ODFs. I am sending several screenshots of their estimation on our testing data. Could you please look at them to asses whether the protocol is tuned reasonably?
WM response functions:
b=0
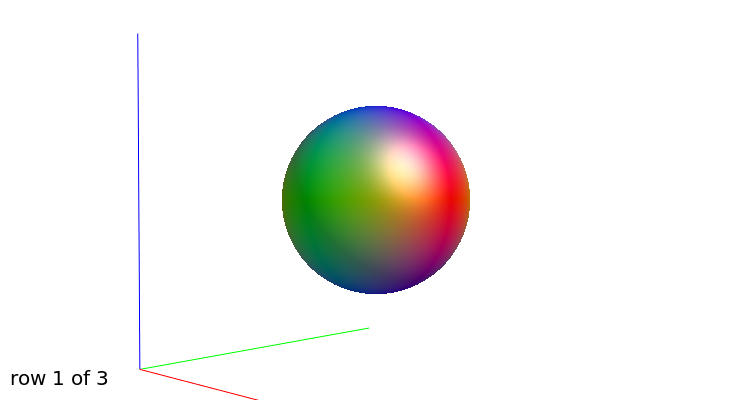
b=1000
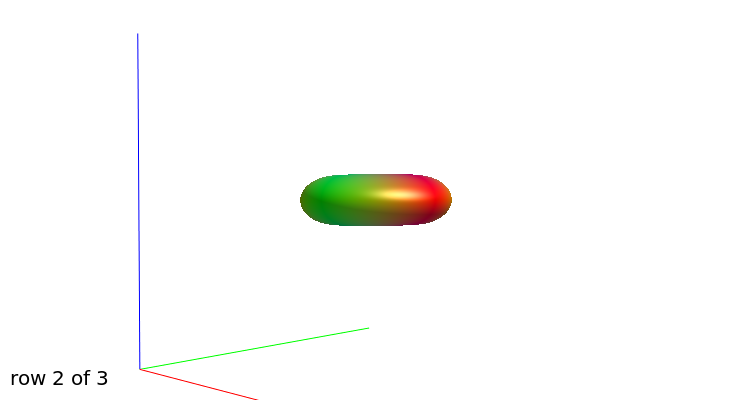
b=3000
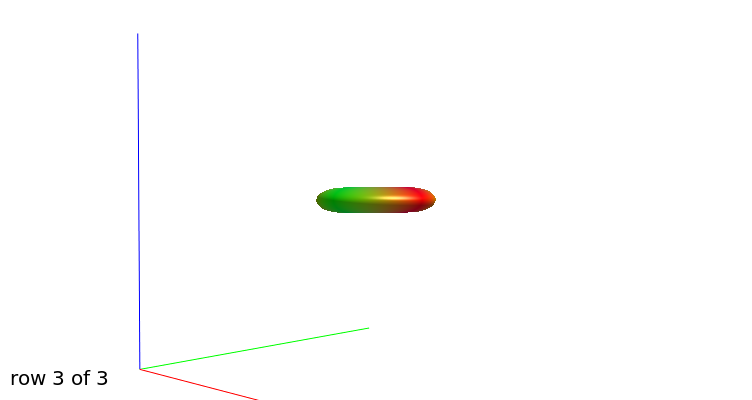
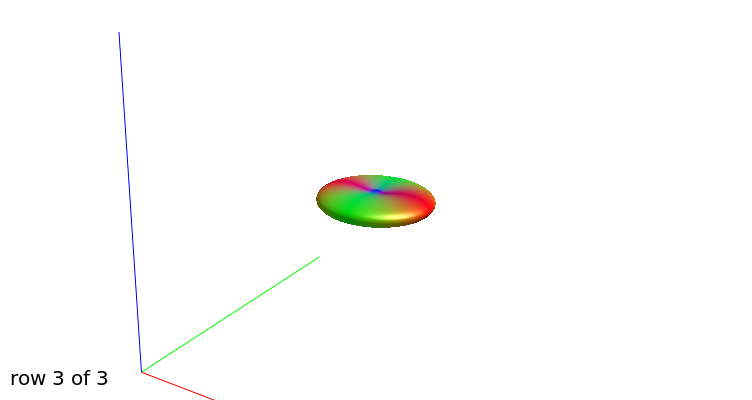
b=3000, another view
FODs in brain stem in region of cerebellar penduncles:


The FOD inspection at focus is showing quite complex shape, at least 3 fibre directions, could it be trusted?
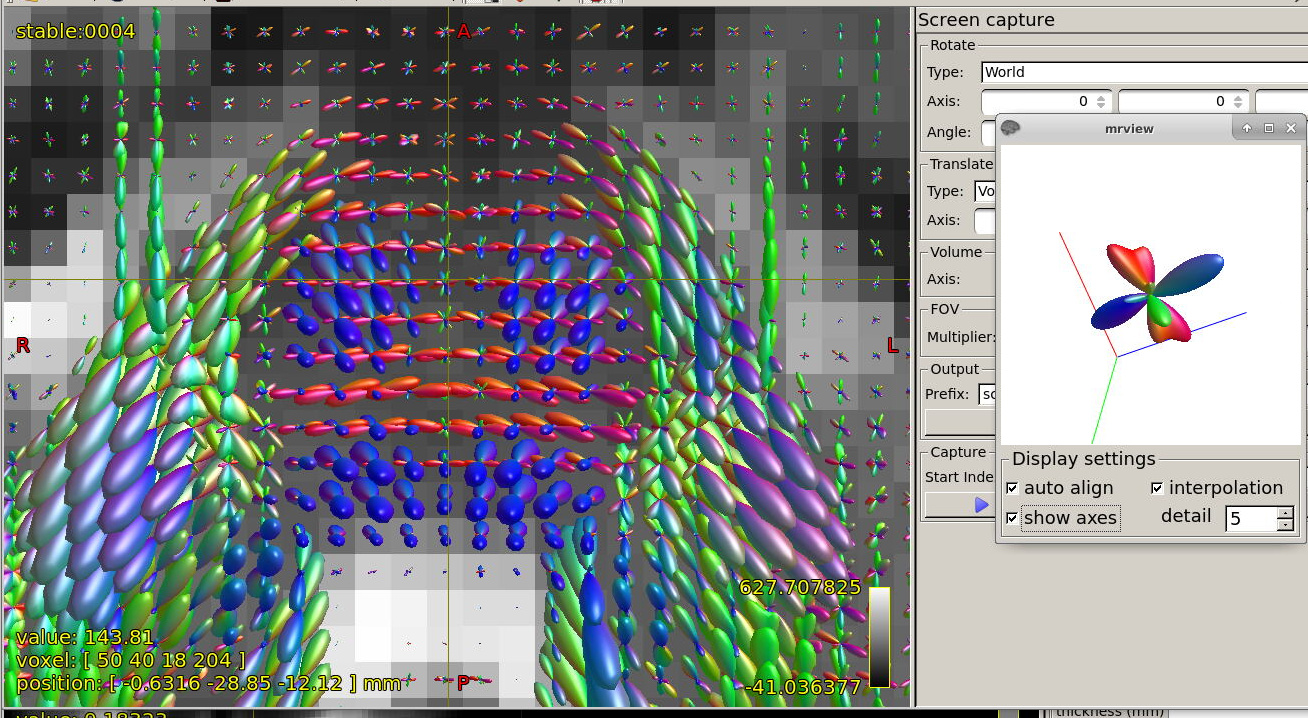
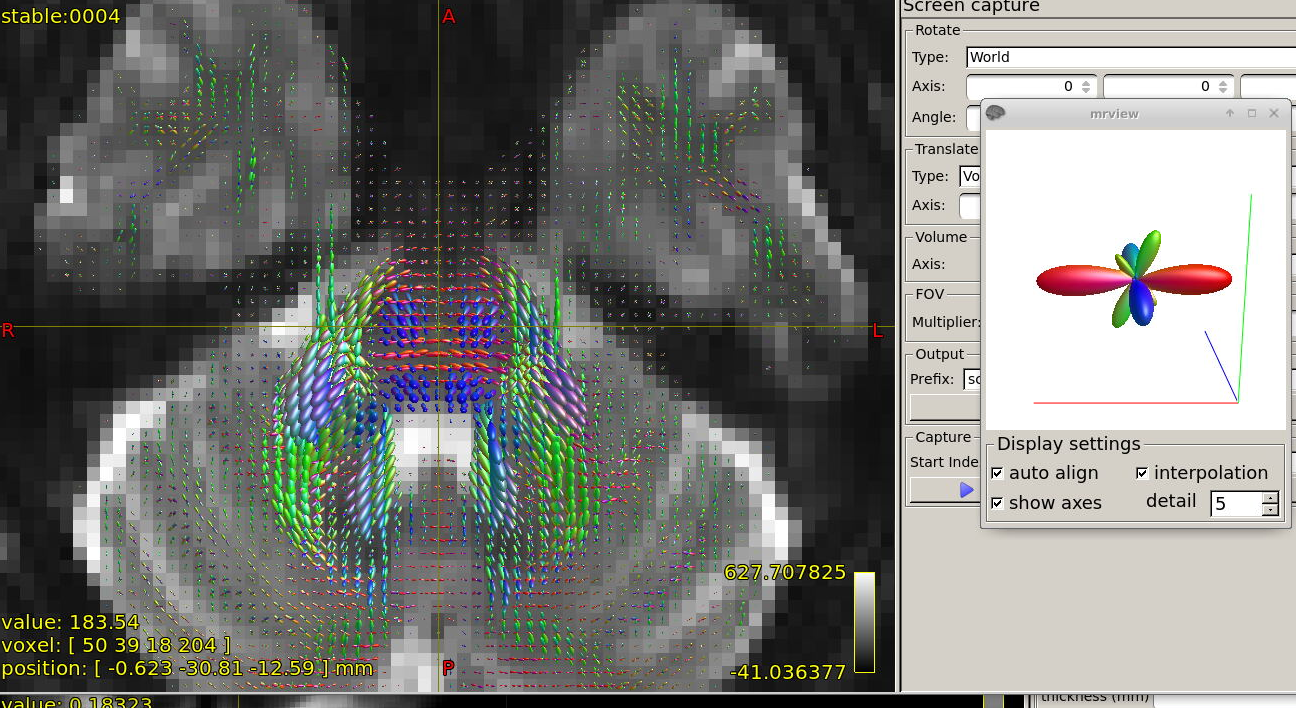
FODs around ventricles:

Could you please comment on? Is this protocol efficiently optimized?
Last questions concerning data acquisition/post-processing:
Do you have experience with the option “dynamic field correction”? I think that it is better NOT to use this, since this correction would be done more efficiently by eddy.
Do you use option to re-scan image when data corruption/image artifact is detected during scan?
Do you use some data filtering on the scanner (image filter)?
Considering post-processing: Would you recommend some upsampling to “gain” spatial resolution?
Thank you very much in advance for your suggestions/comments.
Antonin