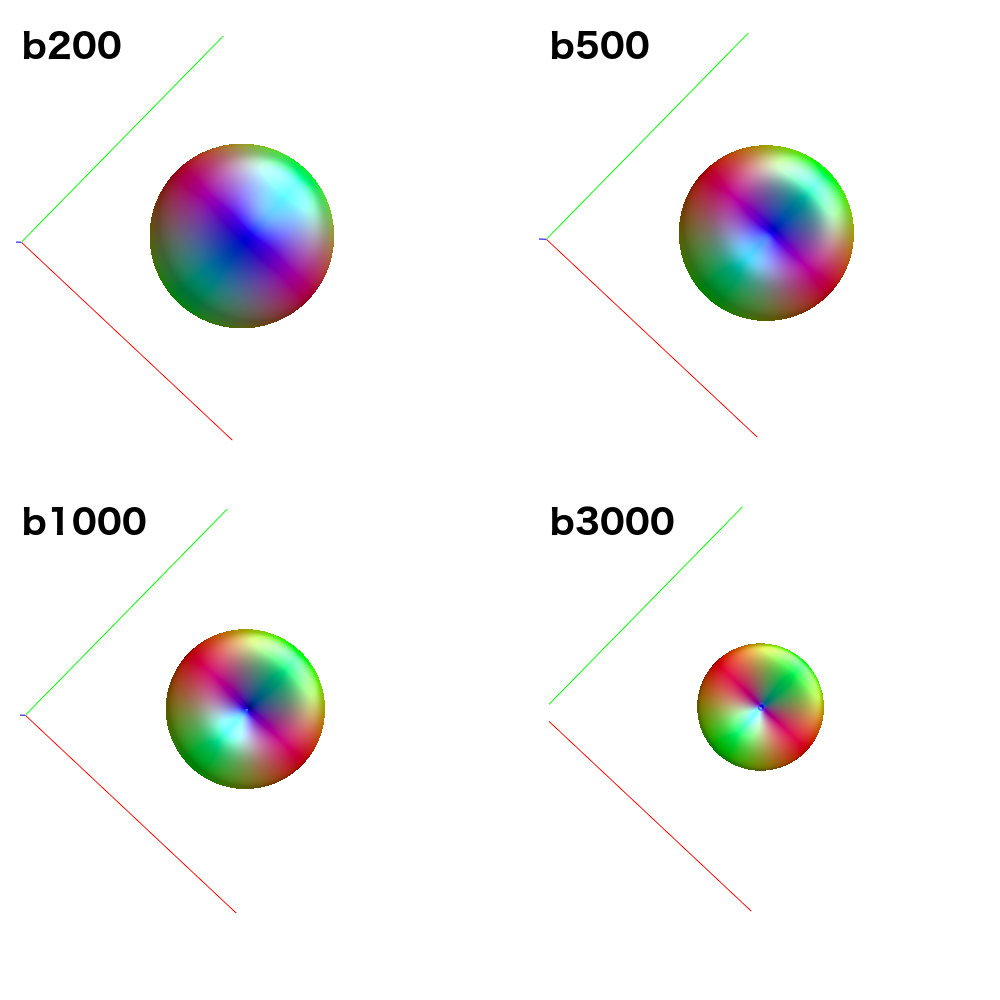
Hey Alistair,
So, a couple of different things to disentangle here.
amp2response
The “golden rule” of having zonal spherical harmonic coefficients decreasing in magnitude (and flipping in sign) with increasing harmonic degree came about from long-term observations of the behaviour of response function estimation, dating back as you say to the old 0.2 version of the software, and makes sense given the expected “pancake” shape of the response function and the reduced signal power in higher harmonic degrees.
This was however based on older methods for estimating a response function from a set of voxels deemed to be “single-fibre”, and the estimated fibre directions in those voxels. The old estimate_response command, as well as the MRtrix3 command sh2response, operate as follows:
Since 3,0_RC3 however, the script algorithms within dwiresponse have been using the new command amp2response (GitHub discussion here; method abstract here). This uses the information from all single-fibre voxels in a single determination of appropriate response function coefficients.
There are two crucial discrepancies here:
-
The amp2response method additionally enforces non-negativity and monotonicity of the response function. The response function coefficients are necessarily modified in order to enforce this constraint.
-
Because it uses data from all single-fibre voxels at once, amp2response can fit a higher harmonic degree than what would have been possible previously. For instance: Your b=200 shell contains 6 volumes; enough for a lmax=2 fit. Previously, this would have meant estimation of a response function with only 2 non-zero coefficients for this shell. amp2response in comparison is providing an lmax=10 response function, despite having only 6 volumes. So while the coefficient magnitudes are not monotonically decreasing, they were in fact previously unattainable.
Now having described that, I’ve no guarantee that estimating and using such high-degree coefficients for low-b shells is actually beneficial; as I flagged in the original GitHub discussion around its inclusion, this may at the very least come off as “unusual” to users, but I’ve never actually had such data in order to test the effect of inclusion of those coefficients (bearing in mind that in the initial transformation of DWI data to SH coefficients for CSD, the SH coefficients of the data in those higher degrees will necessarily be zero). If you felt so inclined, you could experiment to see what kind of response functions you get:
-
Using sh2response instead of amp2response
-
Using the -noconstraint option in amp2response
-
If you really want to get your hands dirty, uncomment this line and re-compile…
Using different shells in dwi2response
Here I need to be very clear in differentiating between the different steps in processing: While amp2response is responsible for taking a pre-determined set of single-fibre voxels and estimated fibre orientations and producing the response function coefficients, the major task of the various dwi2response algorithms is the selection of those single-fibre voxels.
Now I should clarify that given reducing your data to only the b=0;1000;3000 shells, the resulting response function coefficients should correspond to the first, fourth and fifth rows of those obtained when all shells were used. So when you look at the data appropriately, i.e. omitting the second and third rows from the full data results and then comparing to b=0;1000;3000 data, they are in fact very similar. There’s maybe a little numerical discrepancy, which could be due to imprecision in the numerical solver utilised within amp2response, or it could be due to selection of a different set of voxels for response function estimation…
The question then is whether dwi2response msmt_csd or dwi2response dhollander may select a different subset of voxels for response function estimation of each of the three tissues.
-
dwi2response msmt_5tt involves calculation of FA, which is in turn used to refine selection of GM and CSF voxels. But estimation of the WM response function specifically shouldn’t be affected.
-
dwi2response dhollander can really only be clarified with certainty by @ThijsDhollander; I thought it had been commented that it only uses b=0 and the highest b-value shell, but from the current code it seems to be doing some operation over shells, which means the outcome could conceivably change if certain shells are omitted from the data. But I’m fully waving the ignorance flag here.
As mentioned above, it’s possible for there to be some imprecision in the solver used by amp2response, so reporting only variation in those coefficients is not enough information to tell exactly where the variation in results is coming from. So it would be worthwhile using the -voxels option in dwi2response to see whether or not the selection of exemplar voxels is in fact changing.
Rob