Hello everybody,
I want to process some multi-shell neonatal data, following the process that @maxpietsch (and @jdtournier) use in the last ISMRM (Multi-shell neonatal brain HARDI template). I found a similar post here but since was without activity long time, I decided to create this one.
I have neonatal multi-shell data, B0s + 750 (64 dir) + 2500 (64 dir), both shells were acquired in the same session but not in the same acquisition, so I decided to do the following:
For each shell independently: denoising, gibbs ring removal, distortion correction and outlier replacement and bias field correction.
After this I rigid register the lower shell to the average B0 values of the second shell, rotating the bvecs. And finally merging both shells
I decided to do that because the shells were not acquired together, so my first question, is this correct? or I should merge the shells from the beggining and perform the preprocessing together?
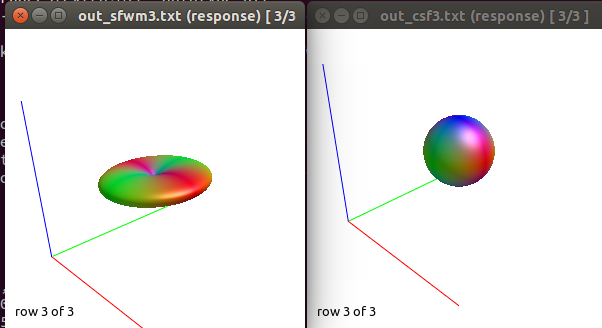
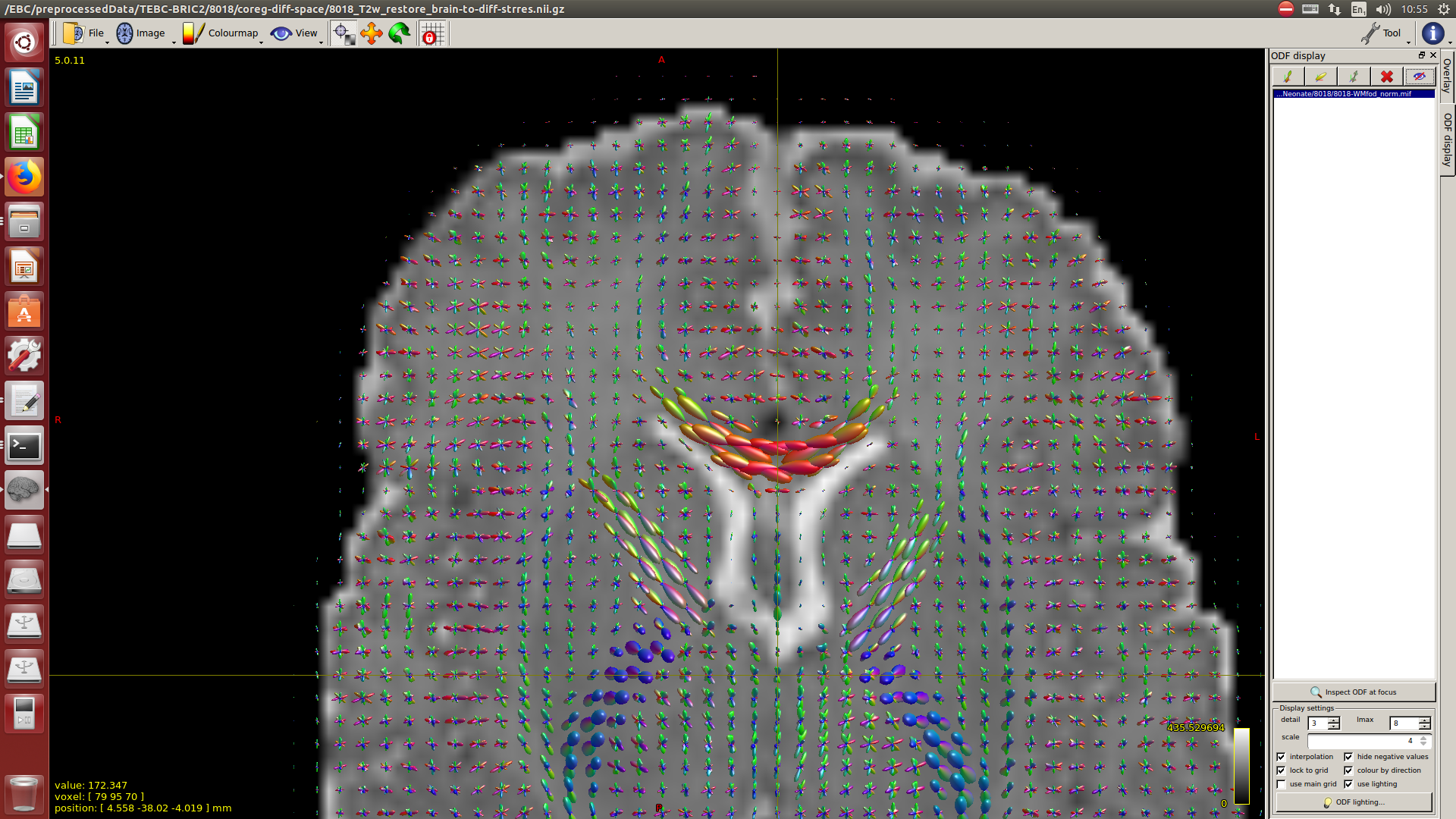
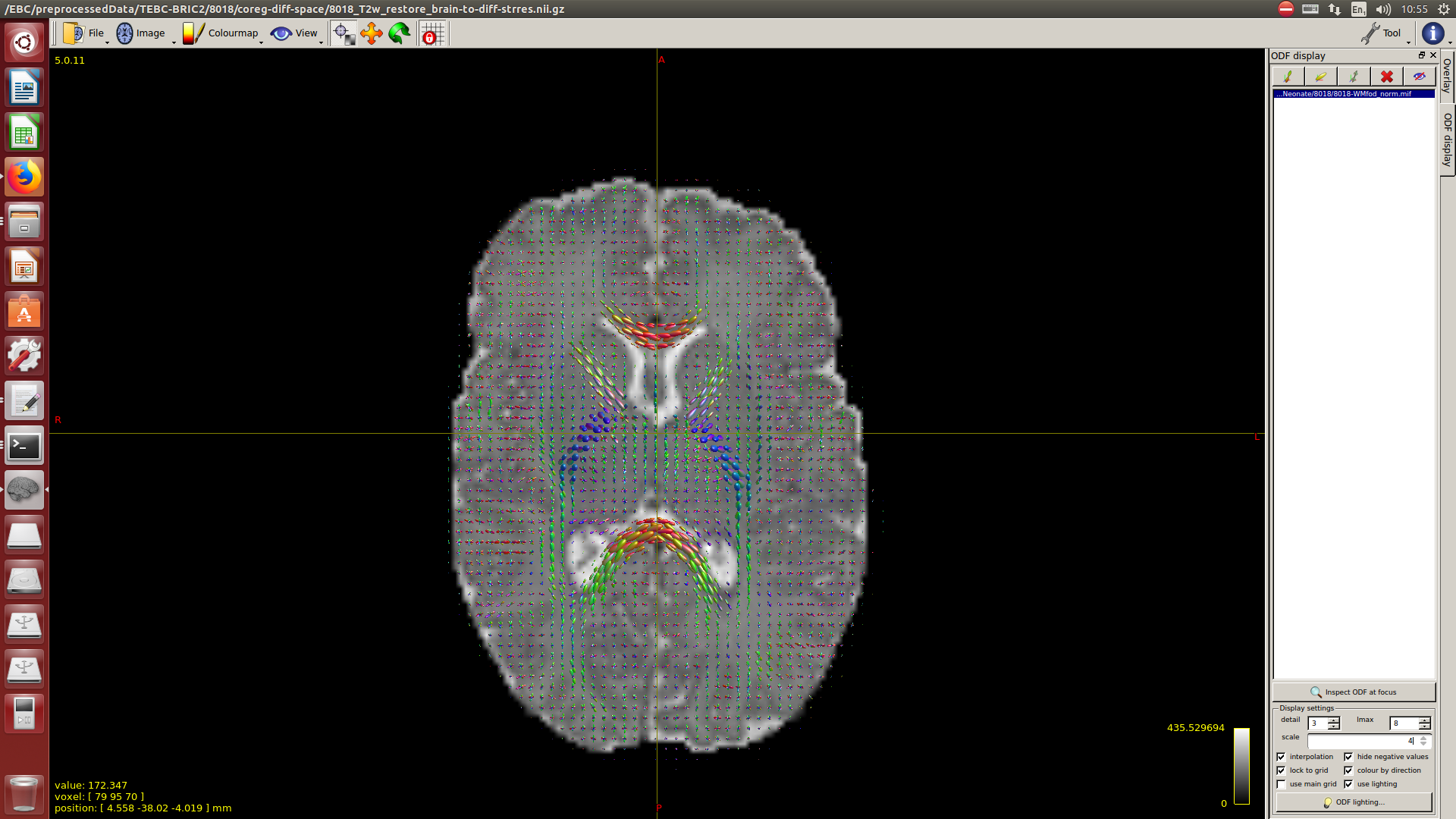
After this, to calculate the WM response function I used the Tournier algorithm in the 2500 shell, and for the CSF response function (because I still don’t have an accurate tissue segmentation for this cohort) I decided to proceed as follows:
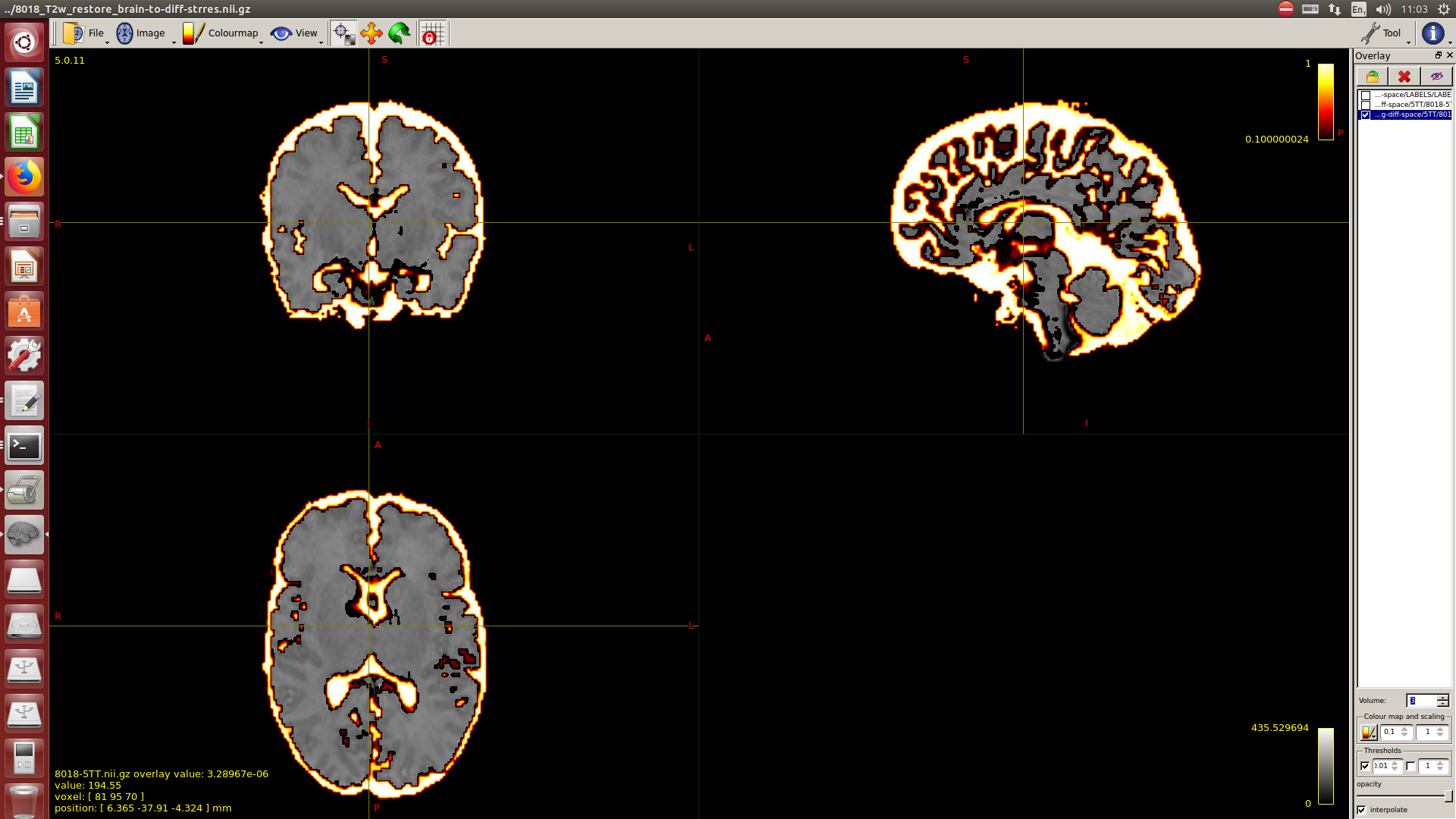
I calculate NODDI maps using AMICCO. The free water was then thresholded to 0.8 and binarized. Then I divided the average 2500 shell to the average B0 shell. I multiply this signal-attenuation imaging for the CSF mask, and I looked for the higher 100 voxels. A mask resulting from this voxels is then used to calculate the CSF response function in the 2500 shells using the Tournier algorithm.
The questions are:
-Does this sound reasonable to you?
-The WM response function looks as expected, but I don’t know what to expect from the CSF function (probably a sphere), how it should be? I was curious to see for example how the CSF response function of the mentioned abstract looks like.
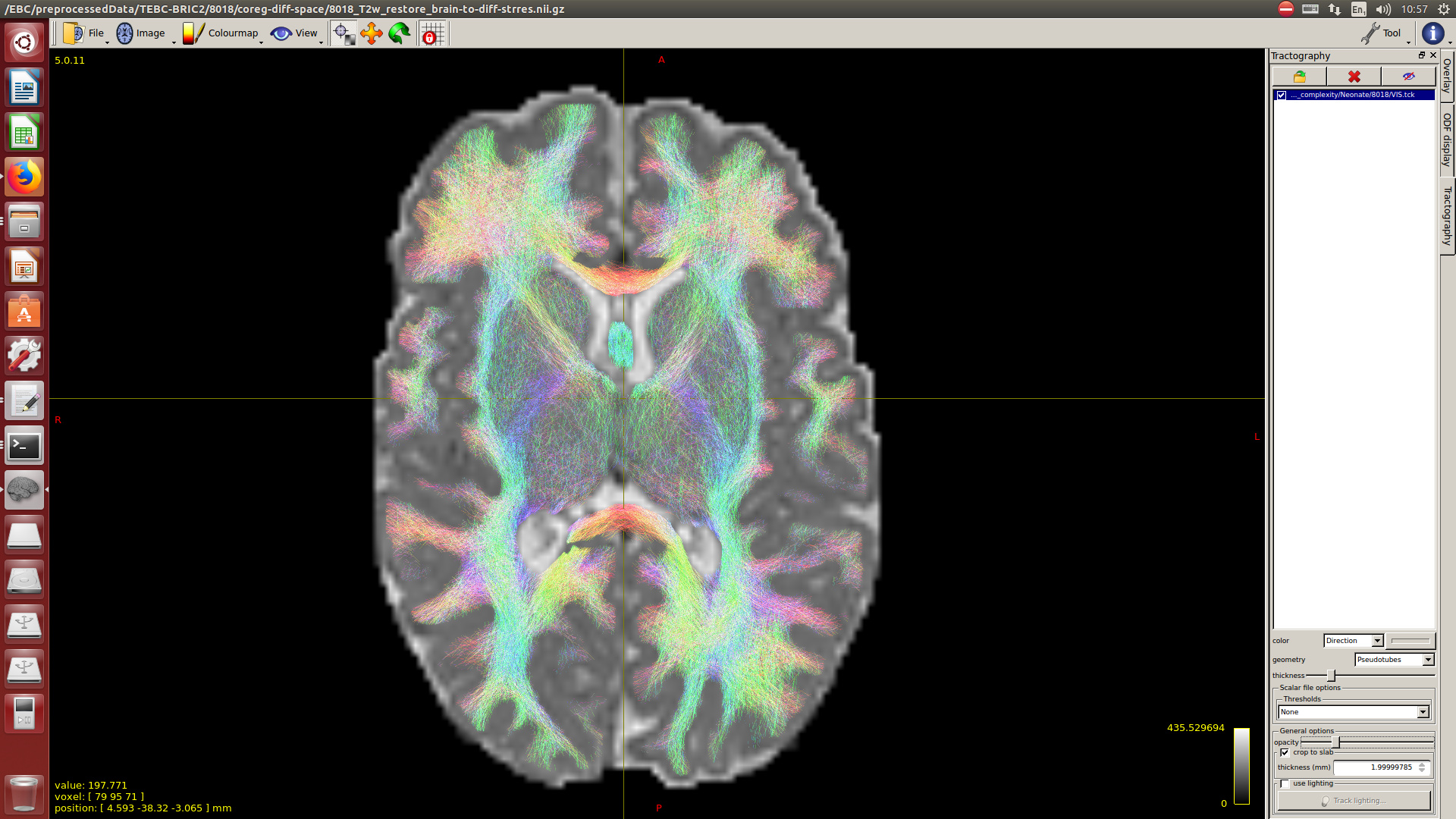
Note that for now, I didn’t perform intensity normalisation across subjects, because for now, I’m interested only in the tractography. But if at some point I decided to add the intensity normalisation and the average response function, how this would affect to the tractography results?
Thanks in advance!
Regards,
Manuel