When I was processing DWI data for a single shell (01000), I found that the final result was very poor. I don’t know what caused it. Can someone help me check the reason?
mrconvert data.nii dwi.mif -fslgrad dwi.bvec dwi.bval -datatype float32 -stride 0,0,0,1 -force -info; alert
date
dwi2mask dwi.mif - | maskfilter - dilate preproc_mask.mif -npass 3 -force
dwidenoise dwi.mif dwi_denoised.mif -mask preproc_mask.mif -noise noiselevel.mif -force
mrdegibbs dwi_denoised.mif degibbs.mif -force
dwifslpreproc dwi_denoised.mif dwi_denoised_preproc.mif -rpe_none -pe_dir AP -eddy_options “–data_is_shelled --slm=linear --niter=5” -force
dwibiascorrect fsl dwi_denoised_preproc.mif biascorr.mif -bias biasfield.mif -force
dwi2response tournier biascorr.mif dwi_response.mif -force
dwi2fod csd biascorr.mif dwi_response.mif dwi_fod.mif -mask DWI_mask.mif -force
tckgen dwi_fod.mif dwi_trck.tck -seed_image DWI_mask.mif -mask DWI_mask.mif -select 100000 -force
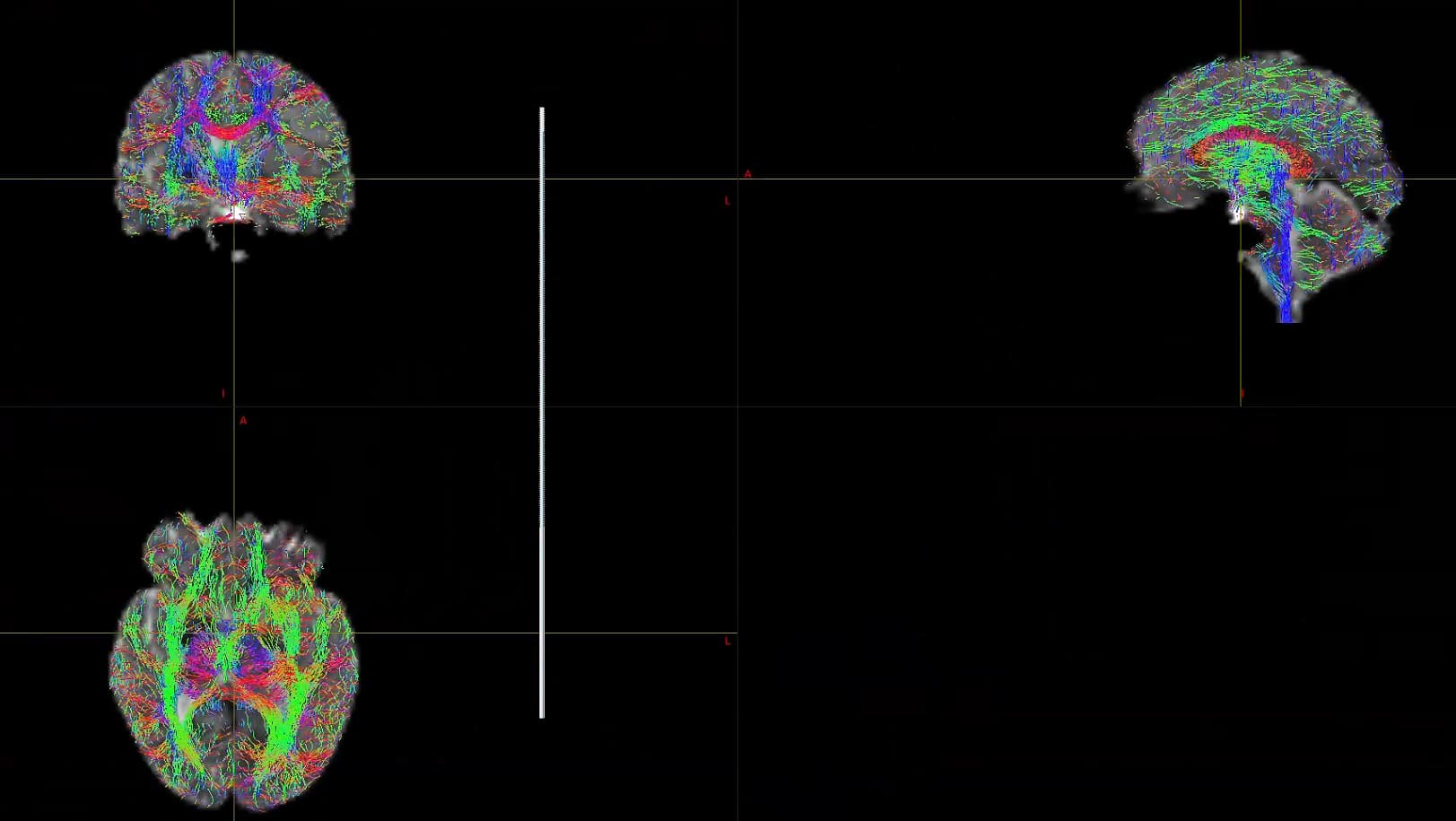
This is the result of my inspection of FOD. From the results, it seems that he did not correctly locate the areas of white matter and gray matter.
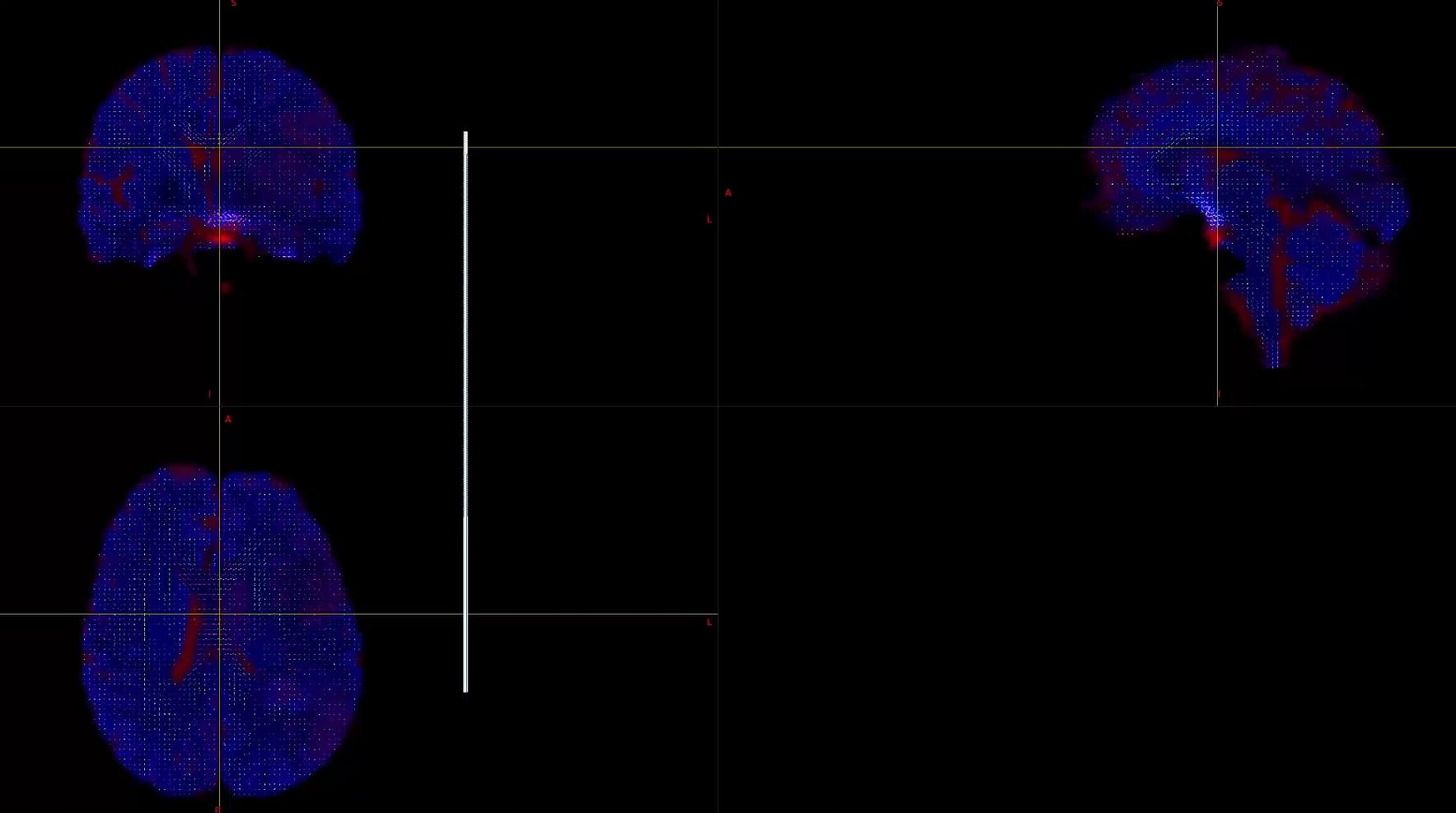
Hi,
Even if only one shell is acquired, still the multishell approach is recommended, using two responses (WM and CSF). Try to replace the following two lines:
By this:
dwi2response dhollander biascorr.mif out_sfwm.txt out_gm.txt out_csf.txt
dwi2fod msmt_csd biascorr.mif out_sfwm.txt wmfod.mif out_csf.txt csf.mif -mask DWI_mask.mif
Another option you have is to use the single-shell 3 tissue algorithm, you can find all the details and a tutorial of how to use it here.
Then use your wmfod.mif as the FOD for your tckgen command. Depending on the usage you want to do with this, remember to use the average response function and mtnormalise. I hope this helps.
Best regards,
Manuel