Hi Ping,
I hope you’re doing well!
There’s a few things that might be going on; though it’s hard to figure out only from the information provided. First and foremost, seeing variation in estimated response functions across datasets can be completely normal if it’s about the general (absolute) scaling of the response functions. So directly looking at those numbers themselves doesn’t tell you much about the variation in shape or contrast between those numbers. It’s the latter that is relevant.
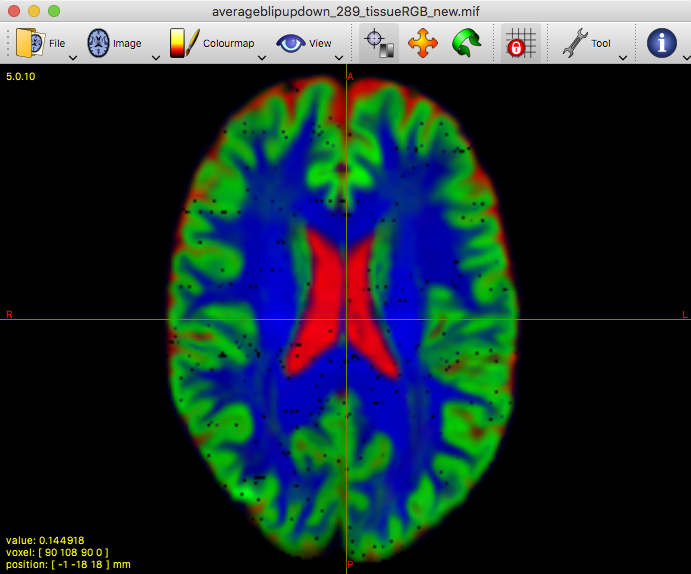
That said, I’ve taken a look at your average WM response function: that looks completely fine actually. Taking into account the successful 3-tissue result you’re showing in the first screenshot using that average response function, I reckon the average WM response function as well as the average GM and CSF response functions are in good condition. Note the latter 2 (GM and CSF) response functions are equally important as the WM response function; they all work together in the modelling result you show in the end!
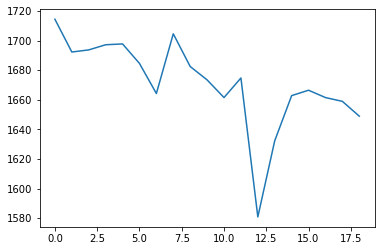
Clearly the second result you show isn’t desirable. Also, that particular individual WM response you show (not the group one) does look different in shape and contrast compared to the average one. Also, even in and of itself it doesn’t look quite right, given certain properties you’d expect for a WM response function.
First things first: aside from your worries about the one individual response function, given your results with the average response functions, I would currently not worry and proceed to use the average WM, GM and CSF response functions without worries across all datasets. The one tweak you could do is to not include the one individual response function you showed into the calculation of the average response function. However, if you’ve got enough subjects, I reckon one individual one will have had very little to almost no impact on the average. So again, I don’t think there’s a great concern of worry per se. Happily continue your analyses in the meantime, I’d say. The other variation, as I mentioned above, is likely only in absolute scale of the response functions. The evidence there is that your average WM response function looks quite good; in a way you’d not expect if the shapes varied too much. So once more, all good I think.
On the side, it’d be nice to figure out what went wrong with the response function estimation of that one individual in particular. Maybe something strange is going on with the dataset itself that you might want to be aware of for example. If that’s the case (e.g. due to some artifact, etc…), there might be reasons to exclude the subject from your study even; so it might be useful to know. Would you be able to share the dataset with me? Feel free to let me know if so, and we can arrange something to make it happen efficiently. In the meantime, it might be handy to mention a few properties about the data: what b-values were used, how many directions, how many b=0 images, etc…? Also, how was the multi-shell acquisition performed: was it all in a single scan, or was it via separate scans for each b-value and did the scans still have to be concatenated in the end? The latter might give rise to some issues that might explain at least some of the things seen in that individual subjects response function.
Cheers,
Thijs
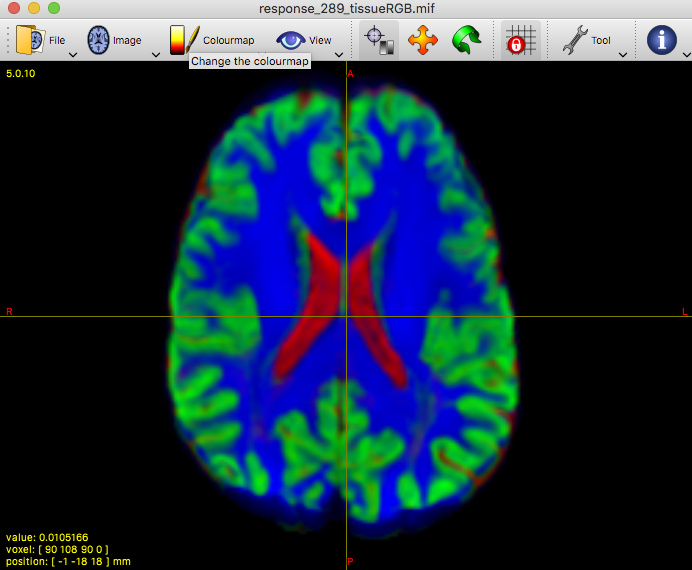
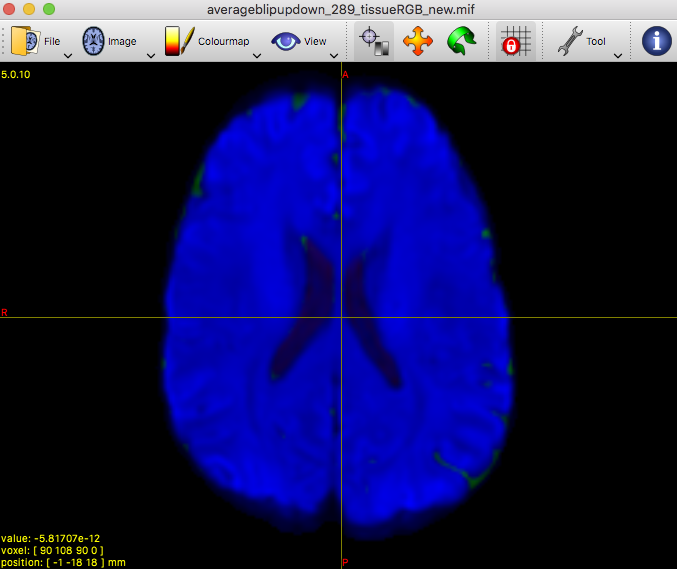
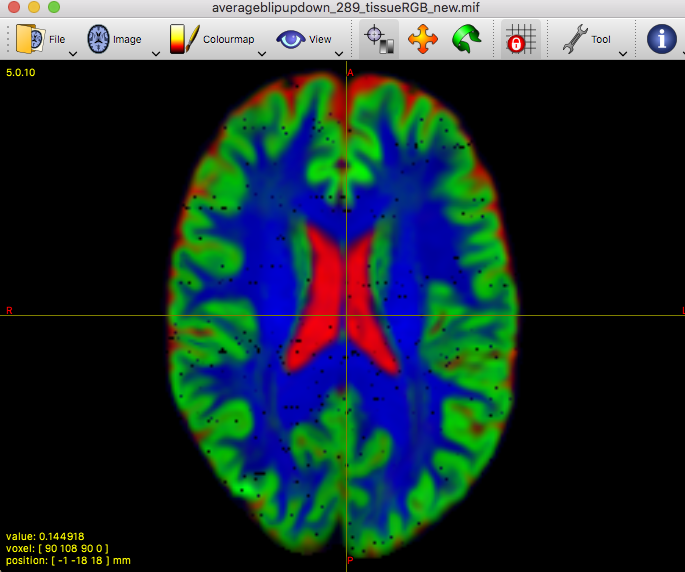

 On the contrary, it allowed to reveal that there must be a problem at least with this particular subject’s data earlier on in the pipeline.
On the contrary, it allowed to reveal that there must be a problem at least with this particular subject’s data earlier on in the pipeline.