Hello, I am a beginner to fiber tracking. I have taught myself the fundamentals using B.A.T.M.A.N and the Mrtrix3 chapter in Andy’s Brain Book alongside the MRtrix3 documentation. However, I find some of the documentation difficult to grasp as a newcomer to this type of data analysis. I likely have some fundamental misunderstandings of this data, so am hoping somebody can guide me in the right direction.
I am attempting to visualise fibers passing through the splenium into early visual areas. Based on my reading of the B.A.T.M.A.N document, my process was the following.
- Generate 10 million whole-brain fibers
- SIFT to reduce this to 1 million fibers
- Generate a splenium mask using ITK-Snap (splenium_mask.nii.gz)
- Use tckedit to create a new .tck file showing only those fibers which pass through splenium_mask.nii.gz (tckedit (tracks_in.tck) (tracks_out.tck) -include splenium_mask.nii.gz)
I have recently been advised to generate 70 million fibers and to use SIFT2 instead to produce tck_weights.txt.
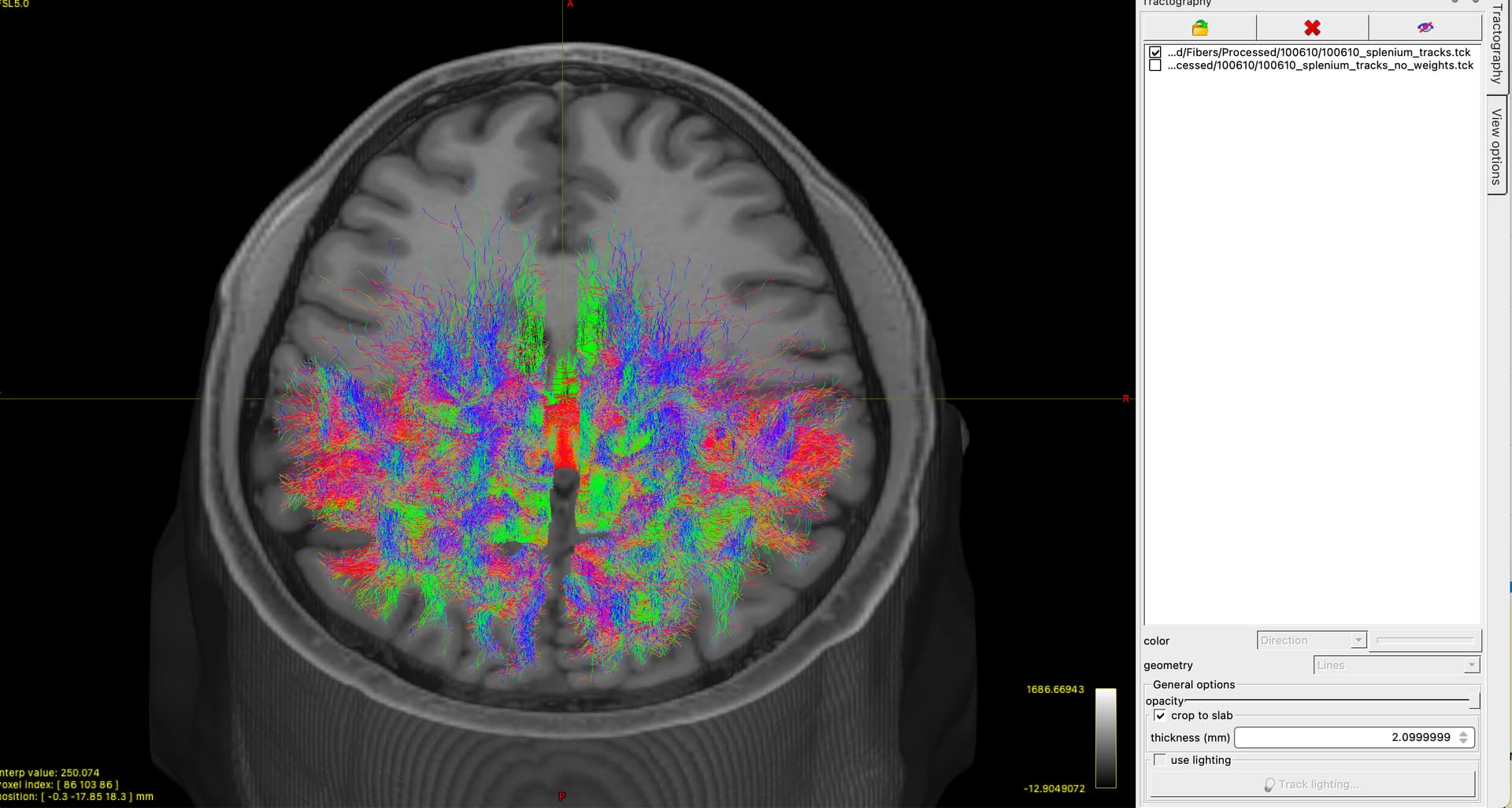
Using the 70 million fibers, I follow the same procedure as above, but add the -tck_weights_in and -tck_weights_out to the tckedit step. To compare, I have generated a track file without these extra options (i.e, no weights/sift applied during the tckedit) and the resulting tracks appear identical. I had assumed adding the -tck_weights_in during the tckedit step would have “filtered” unwanted tracks away.
I understand the weights.txt file simply adds weighting information to the .tck, which does not actually change the file. However, with the updated SIFT2 not actually filtering streamlines down, I’m unsure if this file I have is overestimated or how I should proceed.
I have attached a picture of my output below, and I apologise is this question is full of misunderstanding or beginner mistakes. Thank you for reading, and thank you even more if you’re able to nudge me in the right direction ![]()