Ok, to report back and close this off: we’ve looked into it and solved it offline. The full output until before the earlier reported error was:
dwi2response: -------
dwi2response: 2 unique b-value(s) detected: 0,1000 with 1,64 volumes
dwi2response: -------
dwi2response: Preparation:
dwi2response: * Eroding brain mask by 3 pass(es)...
dwi2response: [ mask: 1499226 -> 1280147 ]
dwi2response: * Computing signal decay metric (SDM):
dwi2response: * b=0...
dwi2response: * b=1000...
dwi2response: * Removing erroneous voxels from mask and correcting SDM...
dwi2response: [ mask: 1280147 -> 1279835 ]
dwi2response: -------
dwi2response: Crude segmentation:
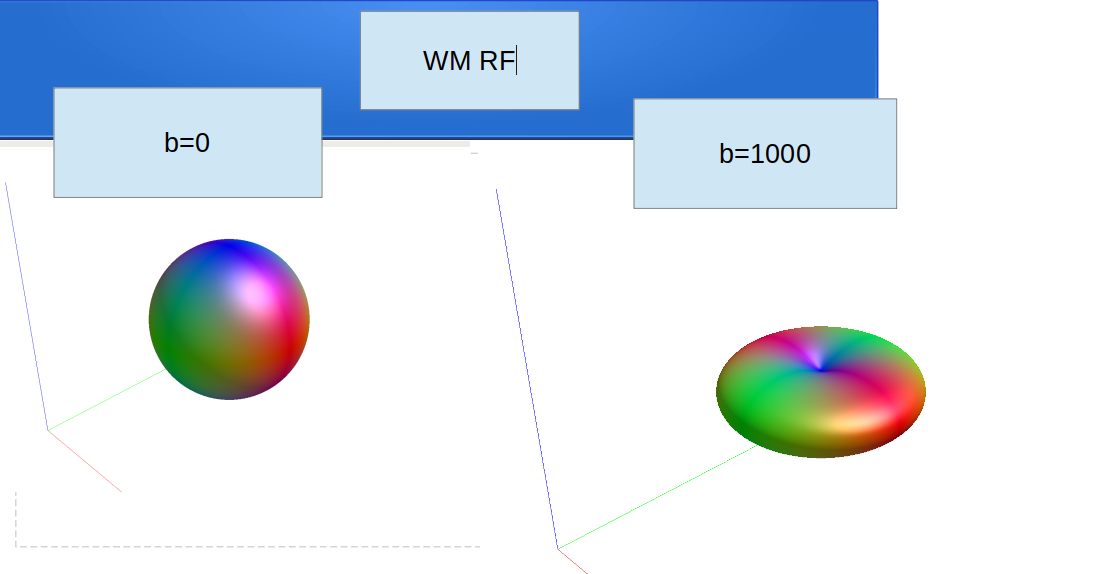
dwi2response: * Crude WM versus GM-CSF separation (at FA=0.2)...
dwi2response: [ 1279835 -> 1279785 (WM) & 50 (GM-CSF) ]
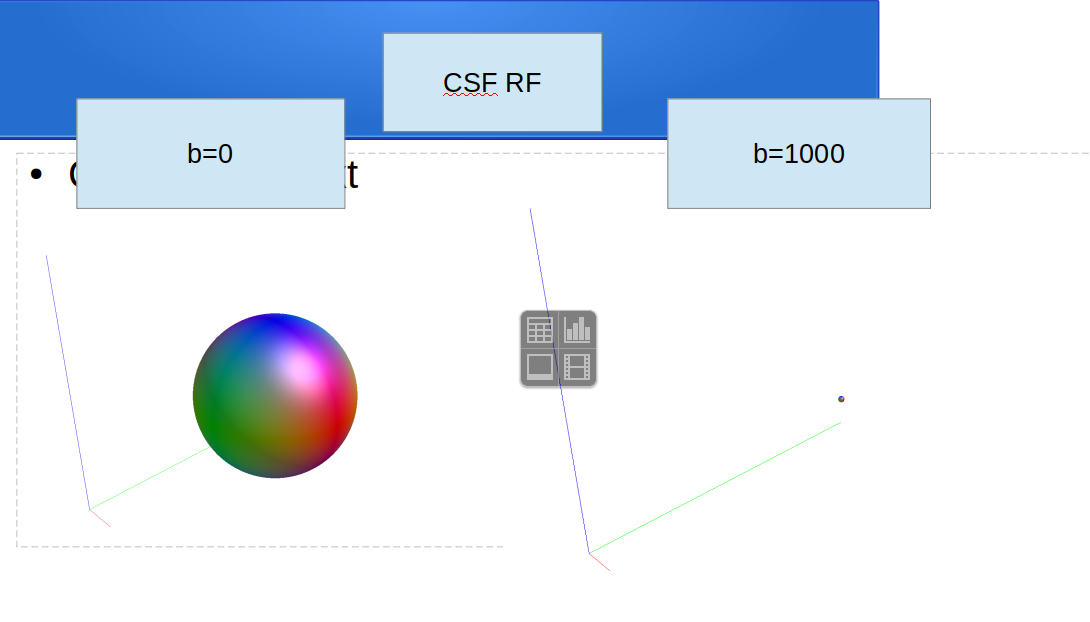
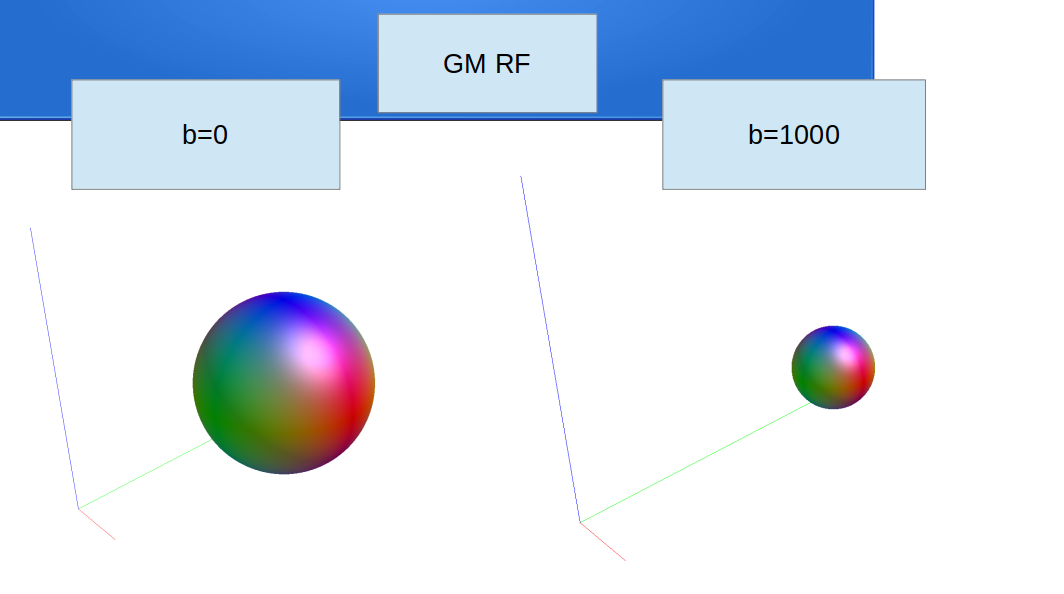
dwi2response: * Crude GM versus CSF separation...
dwi2response: [ 50 -> 17 (GM) & 33 (CSF) ]
dwi2response: -------
dwi2response: Refined segmentation:
dwi2response: * Refining WM...
dwi2response: [ WM: 1279785 -> 1003999 ]
dwi2response: * Refining GM...
dwi2response: [ GM: 17 -> 10 ]
dwi2response: * Refining CSF...
dwi2response: [ CSF: 33 -> 3064 ]
dwi2response: -------
dwi2response: Final voxel selection and response function estimation:
dwi2response: * CSF:
dwi2response: * Selecting final voxels (10.0% of refined CSF)...
dwi2response: [ CSF: 3064 -> 306 ]
dwi2response: * Estimating response function...
dwi2response: * GM:
dwi2response: * Selecting final voxels (2.0% of refined GM)...
…which I could exactly reproduce on my end on the same upsampled data. The data also came with a nice pre-computed mask. No worries there.
The initial clue here was in:
dwi2response: * Crude WM versus GM-CSF separation (at FA=0.2)...
dwi2response: [ 1279835 -> 1279785 (WM) & 50 (GM-CSF) ]
This is the step that separates WM and non-WM crudely initially (which is then later refined, etc…). In any case, the separation here looks very much out of balance, indicating that way too many voxels had an FA value above 0.2. However, looking into the upsampled data itself then, we realised a number of volumes at the end where all zeros. E.g. mrstats of the upsampled data within the brain mask gives:
volume mean median std min max count
[ 0 ] 494.738 388.090057 301.132 -117.253 3791.53 1499226
[ 1 ] 151.67 160.248291 56.3384 -19.7745 863.292 1499226
[ 2 ] 152.909 161.636276 57.8888 -28.6813 802.051 1499226
[ 3 ] 154.691 163.452454 56.8032 -16.6526 912.083 1499226
[ 4 ] 148.126 156.810303 56.2617 -25.557 811.781 1499226
[ 5 ] 154.134 163.085571 56.868 -22.2393 865.215 1499226
[ 6 ] 151.709 159.929688 55.836 -17.5558 851.449 1499226
[ 7 ] 152.727 161.113647 56.5254 -18.3122 897.37 1499226
[ 8 ] 152.41 161.774902 57.0294 -29.8998 868.062 1499226
[ 9 ] 153.687 162.161835 56.4684 -13.26 884.924 1499226
[ 10 ] 149.487 158.265808 56.3392 -17.7466 828.059 1499226
[ 11 ] 149.845 158.797089 56.2865 -14.8034 847.888 1499226
[ 12 ] 149.179 157.764954 56.2122 -15.9005 890.661 1499226
[ 13 ] 152.913 161.815659 55.9334 -18.3439 832.262 1499226
[ 14 ] 146.906 155.168762 55.5958 -29.5223 857.216 1499226
[ 15 ] 149.081 157.652863 55.3762 -5.74525 933.559 1499226
[ 16 ] 148.969 157.750107 55.4795 -23.2521 830.679 1499226
[ 17 ] 150.163 158.642563 56.0775 -7.94573 876.392 1499226
[ 18 ] 154.027 162.568237 56.2677 -11.8833 950.344 1499226
[ 19 ] 150.185 159.171448 57.6265 -24.6021 1002.39 1499226
[ 20 ] 151.181 159.403931 55.9195 -31.5592 881.49 1499226
[ 21 ] 153.933 163.305298 56.899 -29.2348 883.925 1499226
[ 22 ] 154.261 162.684387 56.3751 -30.3851 915.124 1499226
[ 23 ] 149.102 157.824921 55.5241 -26.4253 819.713 1499226
[ 24 ] 153.055 162.450211 56.2618 -23.6808 894.056 1499226
[ 25 ] 156.925 166.234192 56.5952 -18.5176 977.849 1499226
[ 26 ] 152.641 161.784088 57.4353 -24.1844 836.776 1499226
[ 27 ] 150.062 158.706757 55.683 -29.5699 828.574 1499226
[ 28 ] 152.358 161.353195 55.8603 -27.0974 905.059 1499226
[ 29 ] 149.982 159.510071 56.4315 -14.0581 821.621 1499226
[ 30 ] 149.213 158.12854 55.8289 -16.6214 892.409 1499226
[ 31 ] 154.774 163.897568 55.9401 -19.6115 921.185 1499226
[ 32 ] 155.856 164.580597 56.0664 -19.2309 933.737 1499226
[ 33 ] 159.19 168.703522 57.0455 -26.0583 968.283 1499226
[ 34 ] 155.719 165.070358 55.71 -18.1726 897.377 1499226
[ 35 ] 153.395 162.080444 55.4853 -22.4217 868.023 1499226
[ 36 ] 149.216 157.887573 54.7311 -19.353 860.021 1499226
[ 37 ] 145.98 154.940552 55.168 -23.2812 830.195 1499226
[ 38 ] 145.852 154.467316 54.4531 -17.9308 838.842 1499226
[ 39 ] 151.661 160.197876 56.7448 -31.7362 929.559 1499226
[ 40 ] 148.803 156.756592 55.2557 -19.2926 898.824 1499226
[ 41 ] 148.465 157.243225 55.4671 -16.2449 830.806 1499226
[ 42 ] 152.409 161.18512 57.4395 -20.1523 786.045 1499226
[ 43 ] 145.729 153.784531 54.2876 -18.8578 904.257 1499226
[ 44 ] 150.248 159.324127 56.789 -19.3947 907.398 1499226
[ 45 ] 150.504 159.493057 56.1103 -26.5536 875.271 1499226
[ 46 ] 155.168 164.796051 55.9632 -12.6542 879.15 1499226
[ 47 ] 155.245 164.57254 56.7716 -22.087 824.504 1499226
[ 48 ] 148.829 157.496735 55.4244 -27.8474 840.875 1499226
[ 49 ] 71.2541 0 86.0476 -35.6985 866.412 1499226
[ 50 ] 0 0 0 0 0 1499226
[ 51 ] 0 0 0 0 0 1499226
[ 52 ] 0 0 0 0 0 1499226
[ 53 ] 0 0 0 0 0 1499226
[ 54 ] 0 0 0 0 0 1499226
[ 55 ] 0 0 0 0 0 1499226
[ 56 ] 0 0 0 0 0 1499226
[ 57 ] 0 0 0 0 0 1499226
[ 58 ] 0 0 0 0 0 1499226
[ 59 ] 0 0 0 0 0 1499226
[ 60 ] 0 0 0 0 0 1499226
[ 61 ] 0 0 0 0 0 1499226
[ 62 ] 0 0 0 0 0 1499226
[ 63 ] 0 0 0 0 0 1499226
[ 64 ] 0 0 0 0 0 1499226
Volume 49 is already missing data, and volumes 50-64 are all zero. This naturally then caused the artificially increased FA values.
In the end dwi2response dhollander ran without issue on the non-upsampled data. On the upsampled data, it would’ve run alright too if those volumes weren’t zeroed. In general though, I advise people to run dwi2response dhollander on non-upsampled data: much faster, and slightly better designed for it.
So in the end, it wasn’t a problem with dwi2response dhollander, but with the upsampling step. I’ve seen this happen before for others even relatively recently, in particular on various cluster environments (which was the case here too), maybe with complicated network storage setups or file systems, where it ends up failing to write all the output, leaving zeros in the data.