Hi Rob
Thanks for your quick replay. As I find out from your comments, when two unique b-values ( b=0 pluse B>0) are available, two response functions for two tissues ( WM and CSF) can be obtained using dwi2response dhollander. I tested this strategy as follow:
dwi2response dhollander dwi_den_preproc_unbiased.mif -fslgrad sub.bvec sub.bval wm.txt csf.txt -voxels voxels.mif -fa 0.2
but I encountered with the following error massage:
Error: the following arguments are required: out_csf
how can i solve it? I must mention that with running the dwi2reponsd with 3 tissues in the same dwi data, I can get the response functions:
dwi2response dhollander dwi_den_preproc_unbiased.mif -fslgrad sub.bvec sub-40451_session1_dwi1.bval wm_rf.txt gm_rf.txt csf_rf.txt -voxels voxels_rf.mif -fa 0.2
dwi2response:
dwi2response: Note that this script makes use of commands / algorithms that have relevant articles for citation. Please consult the help page (-help option) for more information.
dwi2response:
dwi2response: Generated scratch directory: /media/sn/5EB4CC35B4CC1207/run/sub-40451/dwi/session1/dwi2response-tmp-KLRWGK/
dwi2response: Importing DWI data (/media/sn/5EB4CC35B4CC1207/run/sub-40451/dwi/session1/dwi_den_preproc_unbiased.mif)...
dwi2response: Changing to scratch directory (/media/sn/5EB4CC35B4CC1207/run/sub-40451/dwi/session1/dwi2response-tmp-KLRWGK/)
dwi2response: Computing brain mask (dwi2mask)...
dwi2response: -------
dwi2response: 2 unique b-value(s) detected: 0,1000 with 1,64 volumes
dwi2response: -------
dwi2response: Preparation:
dwi2response: * Eroding brain mask by 3 pass(es)...
dwi2response: [ mask: 171074 -> 129179 ]
dwi2response: * Computing signal decay metric (SDM):
dwi2response: * b=0...
dwi2response: * b=1000...
dwi2response: * Removing erroneous voxels from mask and correcting SDM...
dwi2response: [ mask: 129179 -> 129043 ]
dwi2response: -------
dwi2response: Crude segmentation:
dwi2response: * Crude WM versus GM-CSF separation (at FA=0.2)...
dwi2response: [ 129043 -> 72867 (WM) & 56176 (GM-CSF) ]
dwi2response: * Crude GM versus CSF separation...
dwi2response: [ 56176 -> 39813 (GM) & 16363 (CSF) ]
dwi2response: -------
dwi2response: Refined segmentation:
dwi2response: * Refining WM...
dwi2response: [ WM: 72867 -> 67772 ]
dwi2response: * Refining GM...
dwi2response: [ GM: 39813 -> 23382 ]
dwi2response: * Refining CSF...
dwi2response: [ CSF: 16363 -> 10374 ]
dwi2response: -------
dwi2response: Final voxel selection and response function estimation:
dwi2response: * CSF:
dwi2response: * Selecting final voxels (10.0% of refined CSF)...
dwi2response: [ CSF: 10374 -> 1037 ]
dwi2response: * Estimating response function...
dwi2response: * GM:
dwi2response: * Selecting final voxels (2.0% of refined GM)...
dwi2response: [ GM: 23382 -> 468 ]
dwi2response: * Estimating response function...
dwi2response: * Single-fibre WM:
dwi2response: * Selecting final voxels (0.5% of refined WM)...
dwi2response: Selecting WM single-fibre voxels using built-in (Dhollander et al., 2019) algorithm
dwi2response: [ WM: 67772 -> 339 (single-fibre) ]
dwi2response: * Estimating response function...
dwi2response: -------
dwi2response: Generating outputs...
dwi2response: -------
dwi2response: Changing back to original directory (/media/sn/5EB4CC35B4CC1207/run/sub-40451/dwi/session1)
dwi2response: Deleting scratch directory (/media/sn/5EB4CC35B4CC1207/run/sub-40451/dwi/session1/dwi2response-tmp-KLRWGK/)
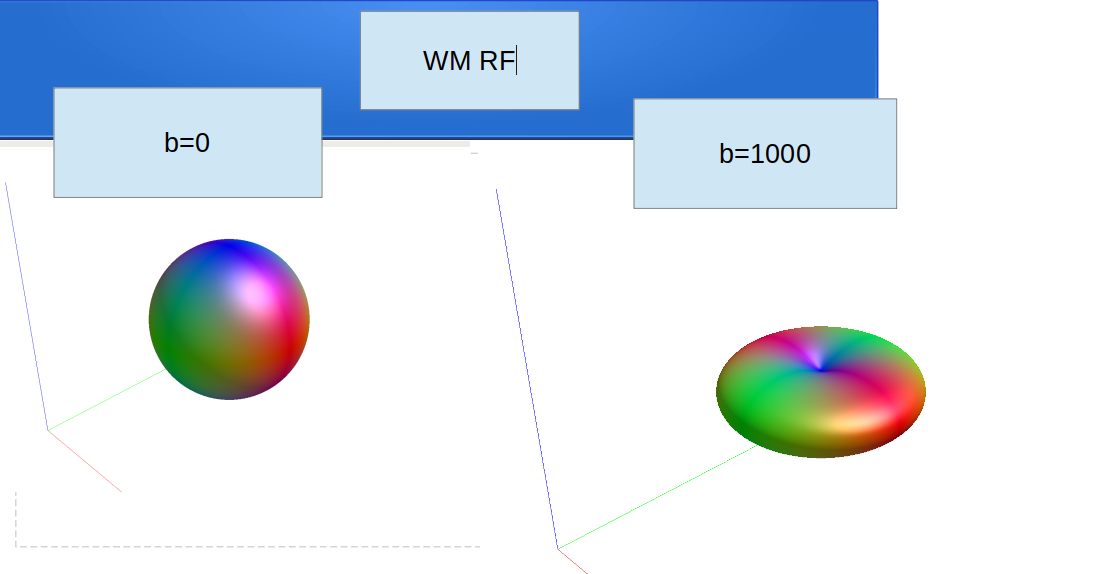
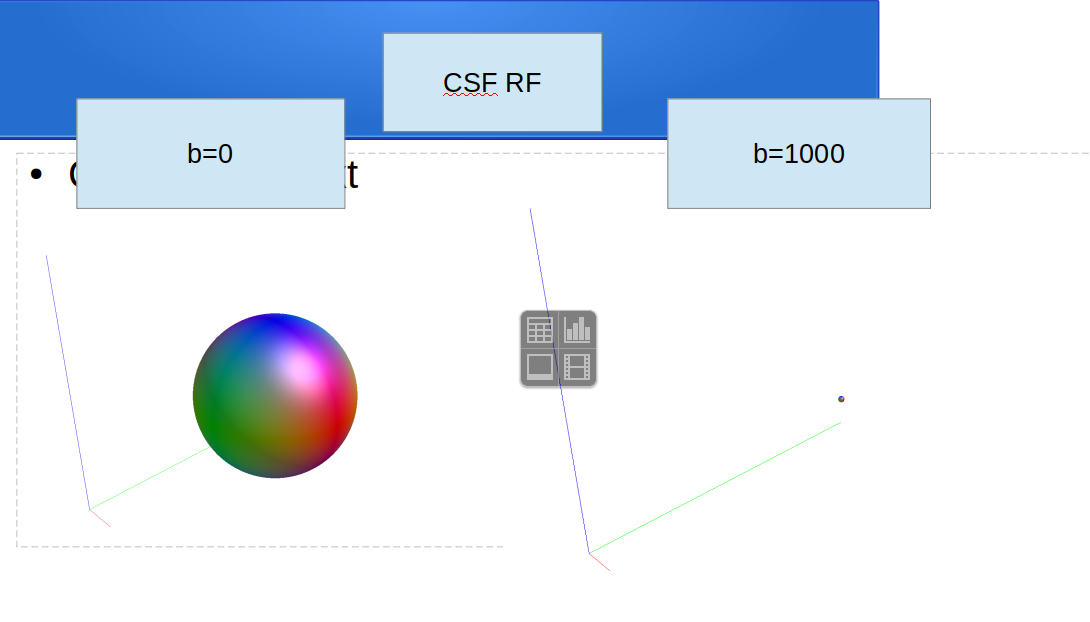
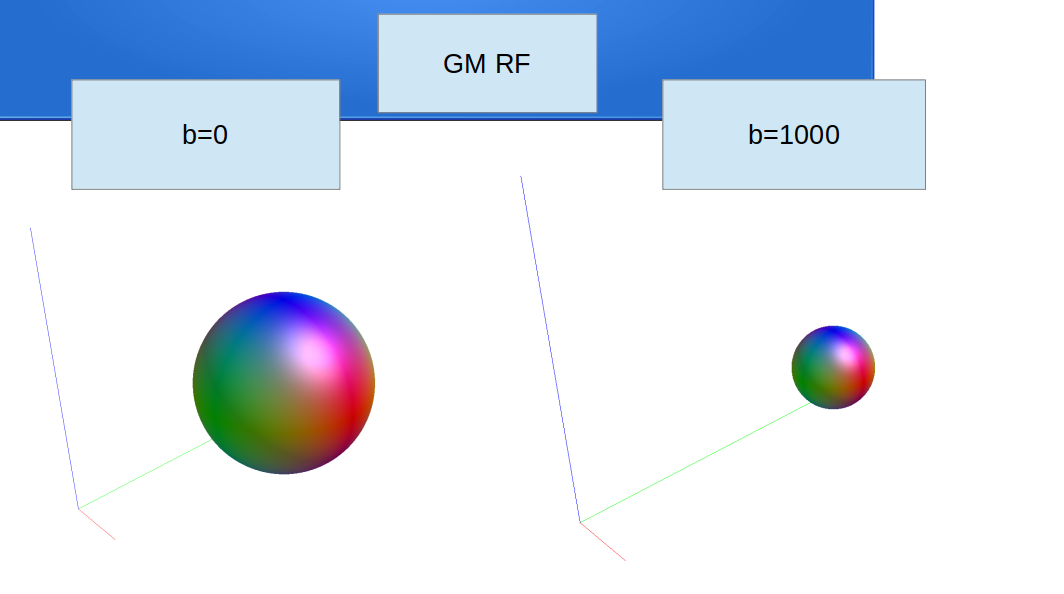
regarding the dwi2fod msmt for single tissue, I use the following codes according to your comments(Fixel-based analysis results - #3 by Andrea_Morelli) for single shell data :
dwiextract -shell 1000 dwi_den_preproc_unbiased.mif dwi_ss.mif
tail -1 wm.txt > wm_ss.txt
dwi2fod msmt_csd dwi_ss.mif -mask mask.mif wm_ss.txt wmfod.mif
Am i doing this step correctly?
bests,
-Milad