Hi @rsmith and @jdtournier,
I have a question about dwigradcheck. I’m running a mouse study and we are collecting a b1000 (30 directions) and a b3000 (64 directions) shell separately but with the exact same settings for each acquisition.
I’m running dwigradcheck because the conversion from Bruker’s format to nifti is wonky and I like to be sure that everything is as it should be.
If I run dwigradcheck on the individual acquisitions, I get one of two options:
-
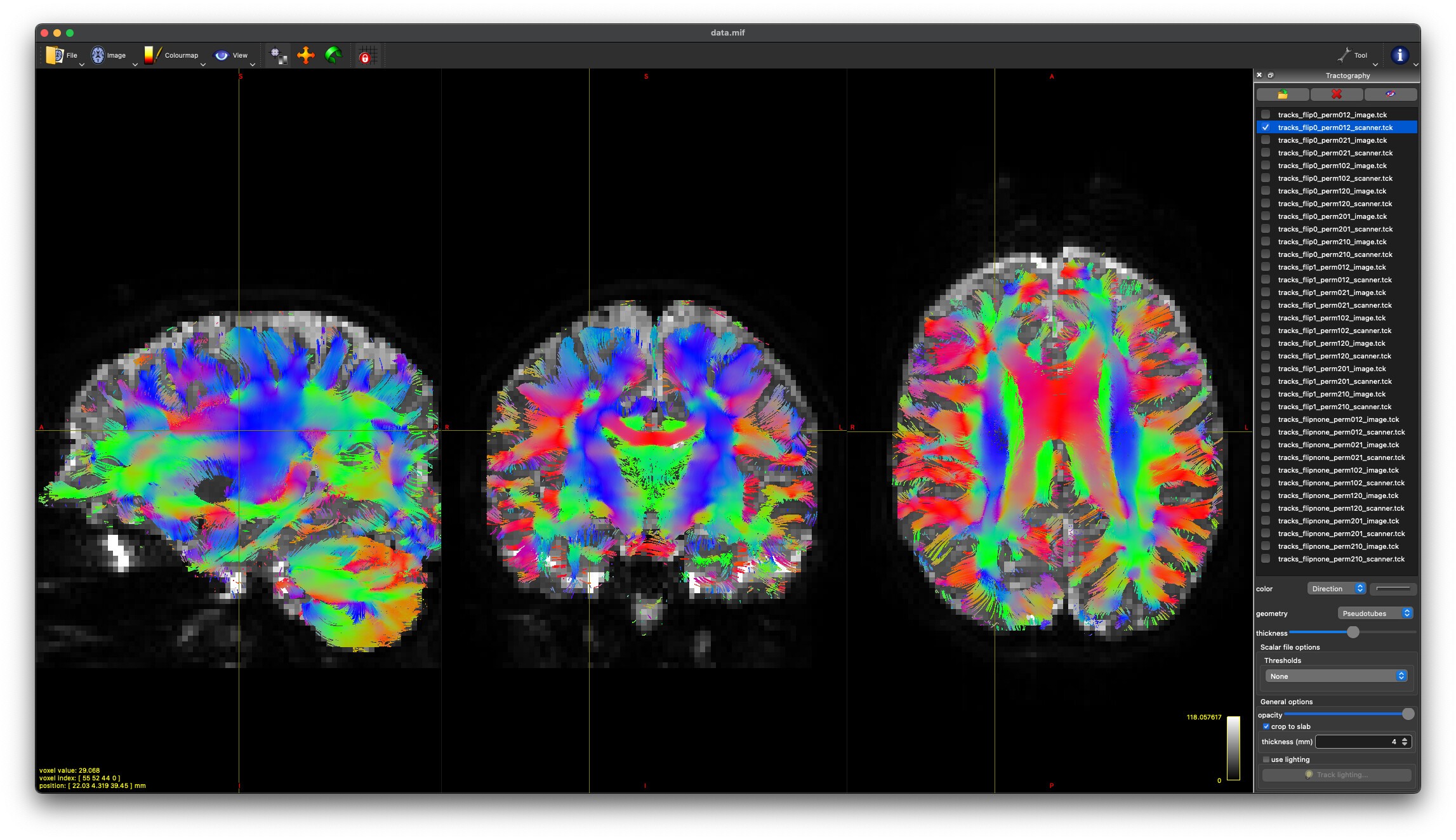
flip1_perm120_image
-
flipnone_perm201_scanner
I see these occur in the same animal (the b1000 will have option 1 and the b3000 will have option 2, for example); however there’s no consistency, like the 3000 always has the flipnone version. Usually the length difference is within 0.05. If I ultimately end up stacking these for eddy and scalars/tractography, is it going to give weird values because of the different flips/permutations/basis? If it is, can I specify which to pick from (since those are the only two selected across the whole dataset)?
Or is this a null point and I should just stack and run dwigradcheck on the combined acquisition?
Hi @araikes,
I reckon @bjeurissen would be best placed to answer this – but I have to agree that converting Bruker data is a hot mess… That said, I still found it surprising that two scans from the same session would end up with different conventions, and that’s enough to give me cause for concern…
One thing that might have a bearing on the convention might be if you are acquiring data with an oblique orientation close to 45°, at which point it may switch from e.g. axial to coronal depending on exactly which side of 45° it happens to fall on. But even that’s a bit of a stretch.
I recommend you have a good look at some simple DTI tractography with these two datasets and double-check that everything looks anatomically correct and as expected in all the major WM pathways. The dwigradcheck script is a great tool, but that’s not to say that’s it’s going to get it right every time…
Cheers,
Donald.
Even with identity transformations, you might get different solutions that are very close in mean length. For instance, we had
Mean length Axis flipped Axis permutations Axis basis
66.66 0 (0, 1, 2) scanner
66.34 1 (0, 1, 2) image
33.27 2 (0, 1, 2) scanner
33.12 2 (0, 1, 2) image
33.03 1 (2, 1, 0) image
32.98 none (0, 2, 1) scanner
32.50 0 (0, 2, 1) scanner
32.48 none (2, 1, 0) image
The tractograms of the first two looked identical to me (check the scratch directory produced by dwigradcheck dwi.mif -export_grad_mrtrix best_grad -nocleanup -number 100000) and I think are equivalent due to the stride order of the input image which if I recall correctly was -3 2 4 1.
The order of the entries can vary if they are very close due to the stochasticity in tracking.
All true. Though that will tend to happen when the images are acquired with little or no angulation – e.g. when the images are almost perfectly axial, and the image and scanner axes pretty much coincide. That’s kind of what we see in your example, @maxpietsch: the top two matches are probably more or less equivalent anyway. What’s weird is that @araikes’s options look like very different permutations of the axes…
1 Like
Thanks @jdtournier and @maxpietsch for your input. I think I’ve solved it (at least in part). A) To your point, these are ex-vivo brains so obliquity is a concern. My testing brain is rotated about 30 degrees to the left. More importantly, B) The software I was using to convert from Bruker to NIFTI made a change in how they were handling the gradient file that is likely incorrect. I reverted it and now for this entire dataset I get two options:
-
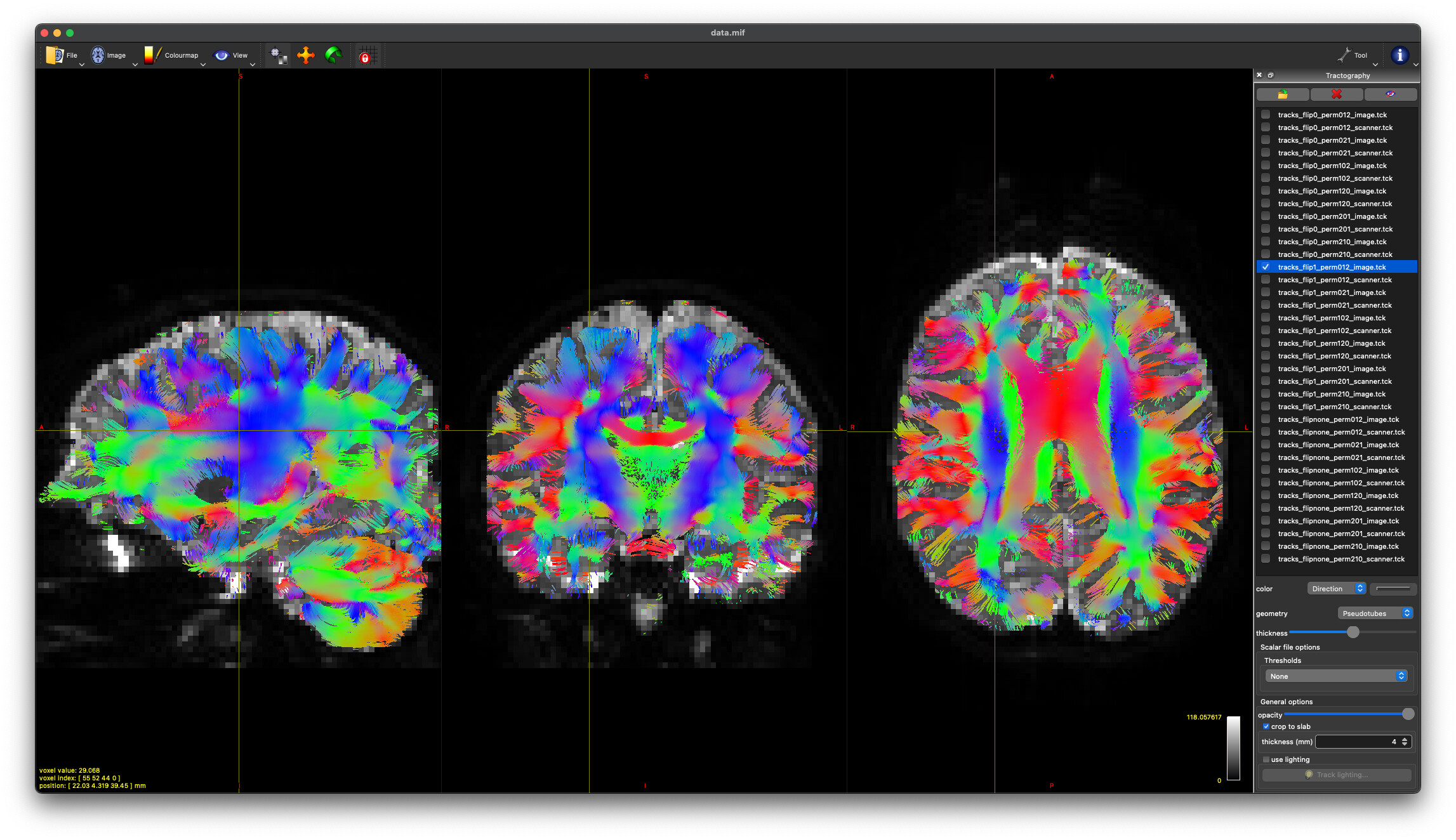
flip1_perm102_image
-
flip1_perm021_scanner
These produce nearly identical tractography (at least visually) with appropriate DEC in white matter regions. And @maxpietsch, thanks for reminding me about the --no-cleanup option. That solves my question about how to pick.
So final question (for the moment)… what is the difference between these two permutations? How does the scanner axis basis differ from the image basis in this context? Is choosing one vs. the other going to make a discernible difference in group-level outcomes?