Hi All,
I am trying to run targeted tractography using a pathway of interest and whole brain tractogram. I generated the pathway between two ROIs and removed the outliers. Then, those final streamlines were merged with the whole brain tractogram with 2m streamlines (after sift). However, after running sift2, the initial fibers can not be extracted with tckedit by using the same ROIs. Besides, the number of extracted streamlines is not equal to the initial pathway. I tried to follow the instruction provided in this post by upsampling the generated tracts, but it did not change the output. Below is the summary of commands and the results:
tckedit wholeBrain.tck pathway.tck combined.tck
(number of streamlines : 2000511)
sift2 combined.tck wmfod_norm.mif -tck_weights_out all_weights
tckedit combined.tck -include ROI1 -include ROI2 -tck_weights_in all_weights tck_weights_out weights_pathway pathway_sift2.tck ( number of streamlines : 359 )
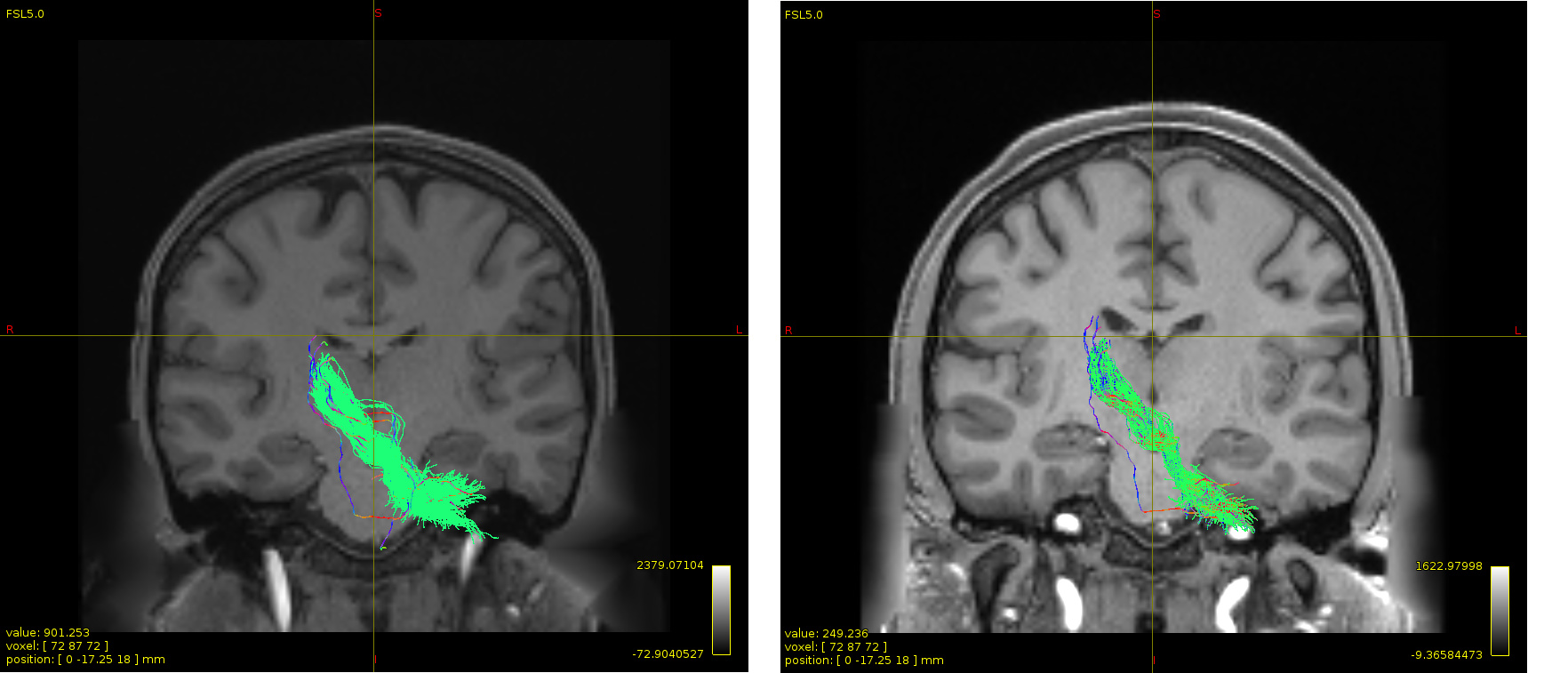
The green color is the tracts after removing outliers that I aim to extract them again after sift2. The other one in the direction-specific color is what tckedit after sift2 generated with some irrelevant streamlines.
tckstats combined.tck
mean median std. dev. min max count
23.4063 10.4075 28.1432 2.46828 125 2000511
tckstats pathway_sift2.tck
mean median std. dev. min max count
102.897 102.496 8.01789 81.0354 123.331 359
Even though I specified the -number option for tckedit, it could not generate the streamlines and gave me the warning message that " User requested 511 streamlines, but only 359 were written to file". Even if it can write the specified number of streamlines in the output file, those are not from the main pathway of interest.
How can I improve the result of tckedit in termes of streamline number and location?
One more question. How does sift2 allocate weights to each streamline in tckedit command? Is it based on one to one correspondence between each streamline in tck file and weighting factor in the text file? In this case, should the providing .tck and txt file be in the same size? I came up with a quick and dirty idea to resolve the problem with unwanted streamlines. I was thinking of running tcksift2 on combined.tck (whole brain tractogram and pathway of interest before removing outliers) first and then extracting the pathway with the output weights (for instance, 1900 tracts). Following that, outliers should be removed to get the final pathway (e.g. 500 tracts). To create weights for new pathway, I need to rerun tckedit.
tckedit newPathway_OutliersRemoved.tck -include ROI1 -include ROI2 -tck_weights_in Pathway_weights_beforeRemovingOutliers -tck_weights_out finalweights
However, with this approach, the input weights file (1900 weights) is larger than the new .tck file. I tested it. It created the output weights file, but I don’t know whether it computes correctly. For example, if it allocates the first 500 weighting factors to the output file, then it doesn’t reflect the correct weights. Am I right?
Cheers