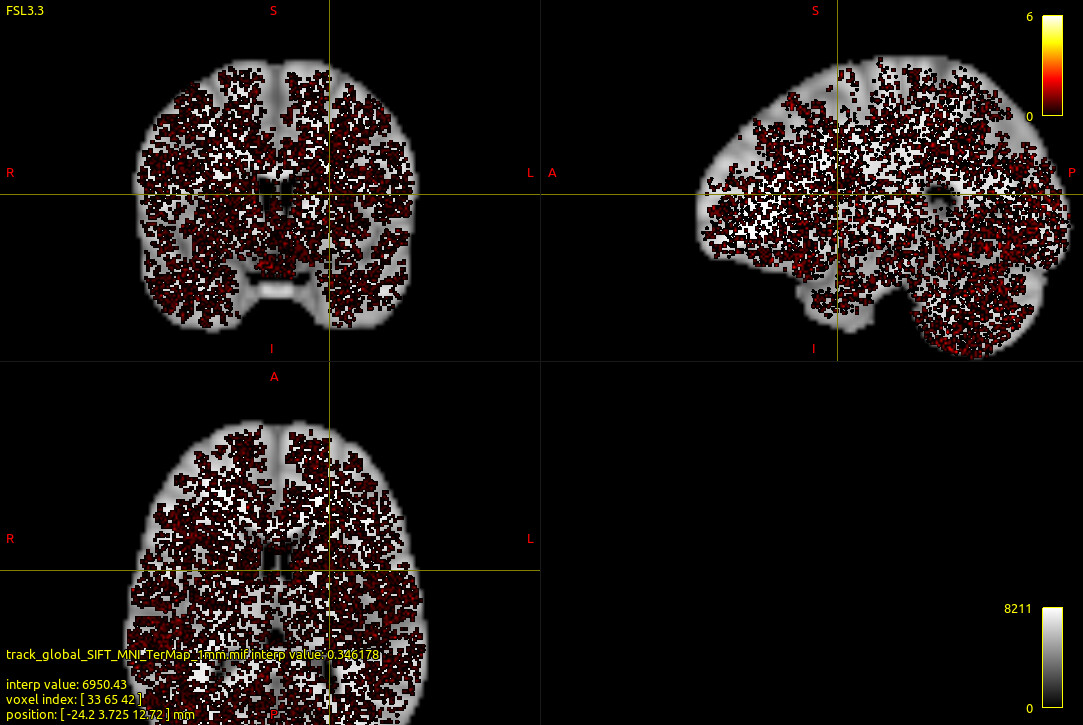
I’m using single-shell data to perform whole-brain tractography. I have tried three algorithm,SD_STREAM , iFOD2 and MSMT global tractography. But the streamline length got from global tractography is very short. I calculated the terminal map and the terminal of steamline diftrubuted throughout the brain.I think that’s not right.
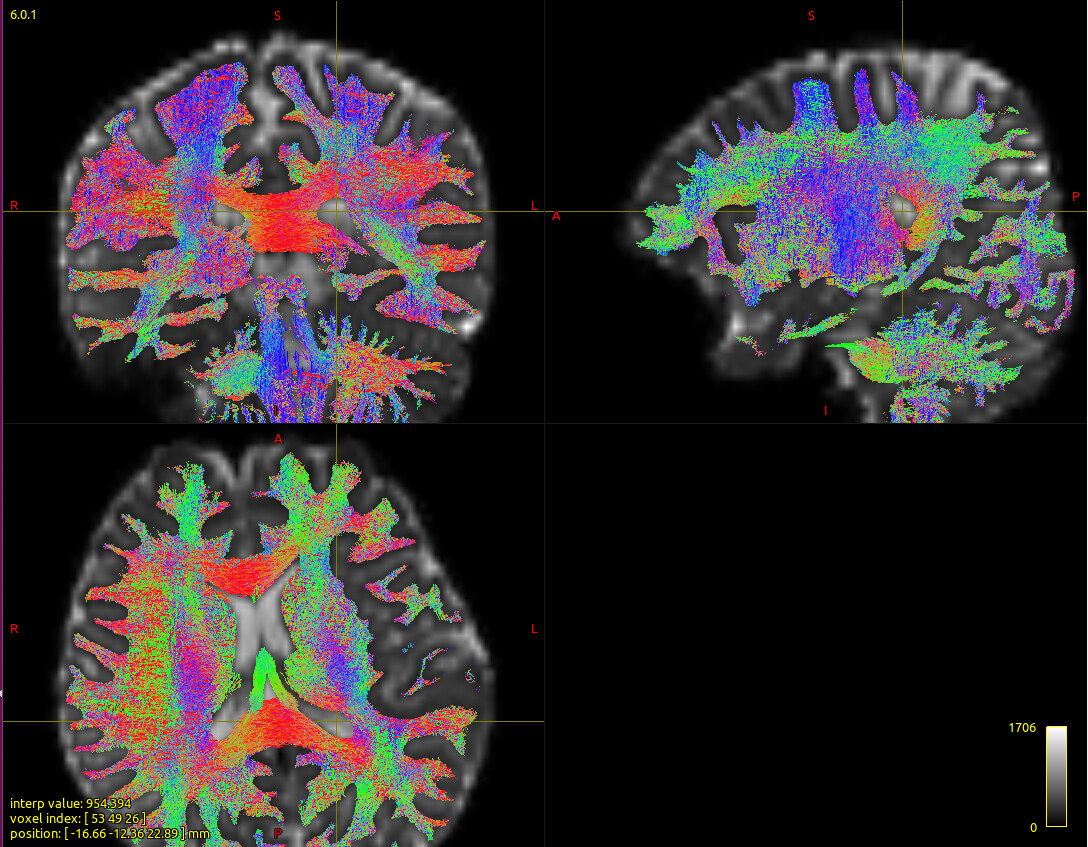
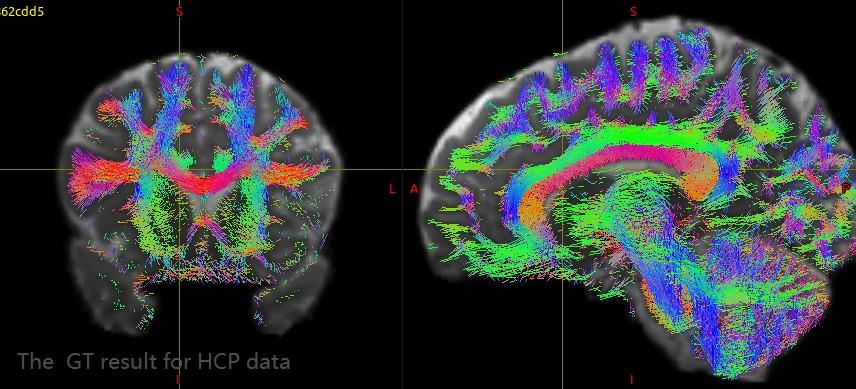
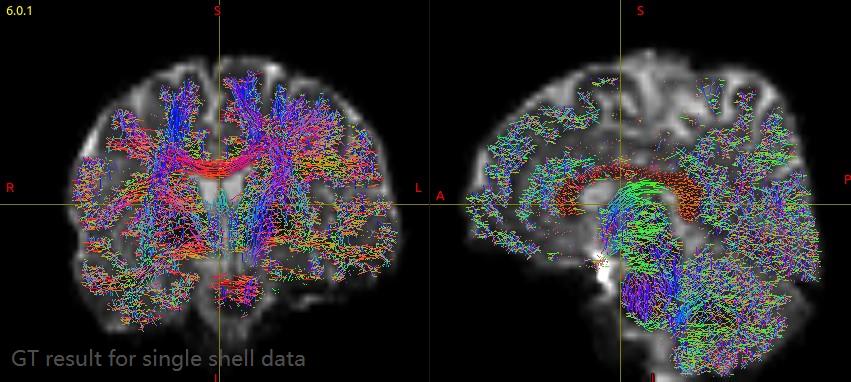
The streamline obtained by golbal tractography algorithm and terminal map are shown in the figure below:
Steamline obtained by global tractography was transformed to MNI space. Then the terminal map was calculated and overlaid on MNI T1 image.
The data was scanned on 3T scanner. There are 64 diffusion gradient directins with b-value=1000 and one unwighted volume. Voxel size is 2x2x3mm.
The scripts I used are as follows:
For global tractography, I used WM mask and ditch the GM response according to ThijsDhollander’s answer and all the parameter are default.
Am I using the right parameters? Or do I need to adjust the parameters?
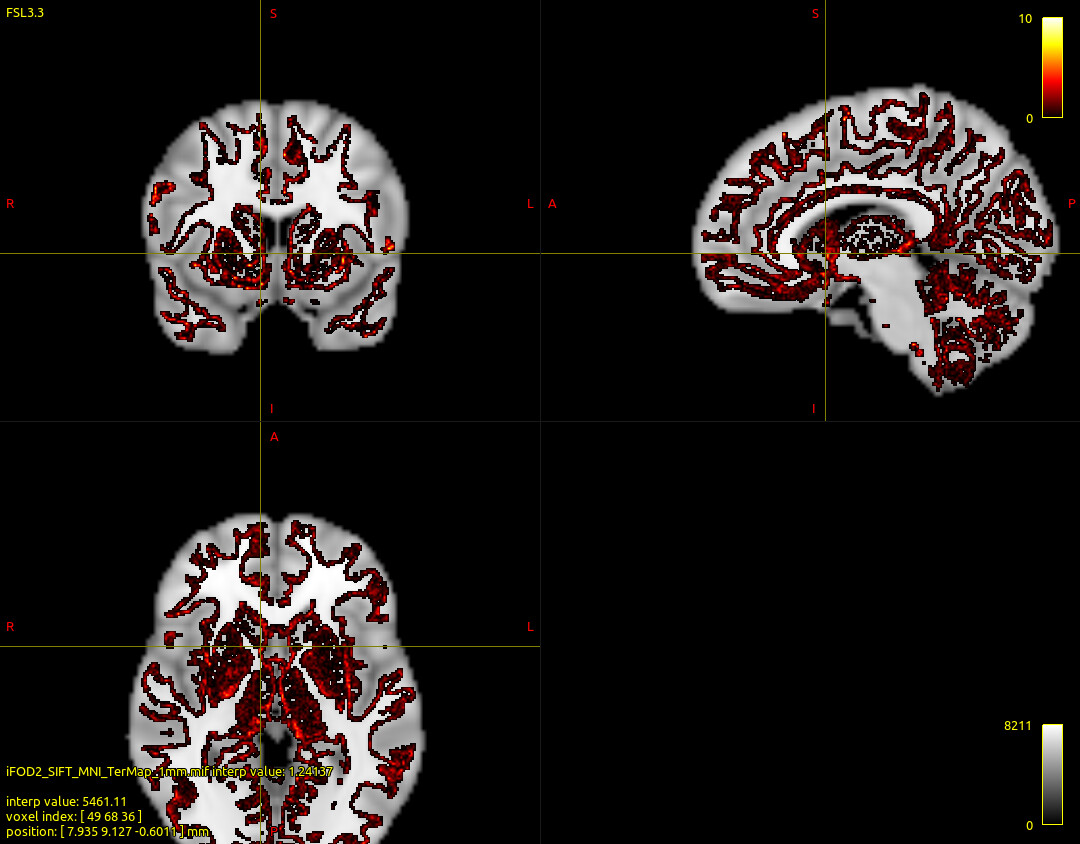
In addition, I also performed whole-brain tractography with iFOD2 algorithm. I used ACT and SIFT during tracking. The results are better than global tractography. Whether the terminal map results are incorrect is related to the absence of ACT? Can ACT be used for global tractography?
iFOD2 tractography command and results are as follow:
It’s not uncommon for global tractography to require some tweaking of parameters to get reasonable results. I’ll leave to @dchristiaens to make any particular suggestions (perhaps after ISMRM deadline).
I calculated the terminal map and the terminal of steamline distributed throughout the brain.I think that’s not right.
You’re correct in that it’s “not right”; where I think you may be misunderstood is in thinking that this is somehow specific to your data or usage, when in fact it’s a long-standing, under-reported dirty secret of almost all global tractography algorithms. There is internally an energy trade-off between wanting to join segments to form longer chains, and the inevitable poorer fit to the image data that results. Parameters can be modified and annealing strategies employed to mitigate it, but you’re unlikely to improve the issue by leaps and bounds.
The only method I would classify as genuinely “global” that addresses this issue is this one. It comes with its own assumptions and limitations, but it addresses the specific requirement of trajectories terminating in the GM.
Whether the terminal map results are incorrect is related to the absence of ACT?
Correct.
Can ACT be used for global tractography?
There is no trivial “extension” of ACT to what I call “segment-wise” global tractography algorithms (the article linked above I refer to as the sole “fibre-wise” global tractography algorithm). These algorithms by design initially contain segments that terminate within the WM, and ACT by design operates in a strictly local step-wise fashion, so they are fundamentally incompatible.
If you re-express the question as “Can one use tissue segmentation information to constrain the final result of global tractography”, the article linked above is the only one I’m familiar with myself; to do this within a segment-wise global algorithm, while I think technically feasible, does not to my knowledge yet exist.
Thank you very for your reply , and learned a lot about global tracking from your reply.
By the way, I’m look forward to some other advice. @dchristiaens
I think Rob has already said the main point: global tractography can not trivially enforce anatomical constraints in the way that ACT does. It is therefore not surprising to find track endpoints in WM with global tractography. Streamline tractography without ACT would also lead to occasional track endpoints in WM.
The situation is exceptionally bad in your case because you are using single-shell data with a fairly low b-value to boot. Multi-shell multi-tissue GT is designed for multi-shell data, especially with default parameters. Using, as you have done, a WM + CSF response function pair is probably the best way to go in such data, but the result will always be sub-par to a certain degree.
You may be able to improve the results slightly if you raise the particle potential -ppot a bit (try e.g. 0.1) to increase the cost of adding a segment, and/or if you raise the connection potential -cpot (try e.g. 1) to increase the reward of connecting segments to one another.
I changed the pararmeter and processed single shell data and HCP data again. The HCP data GT results have a great improvement, but the results of single shell data are still not very good.
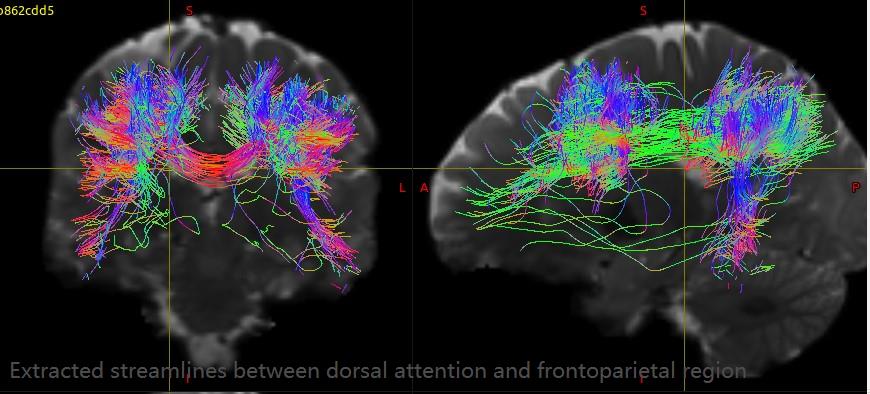
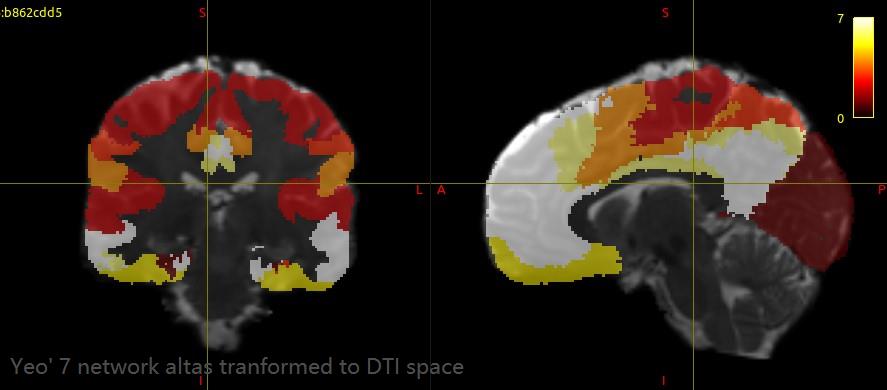
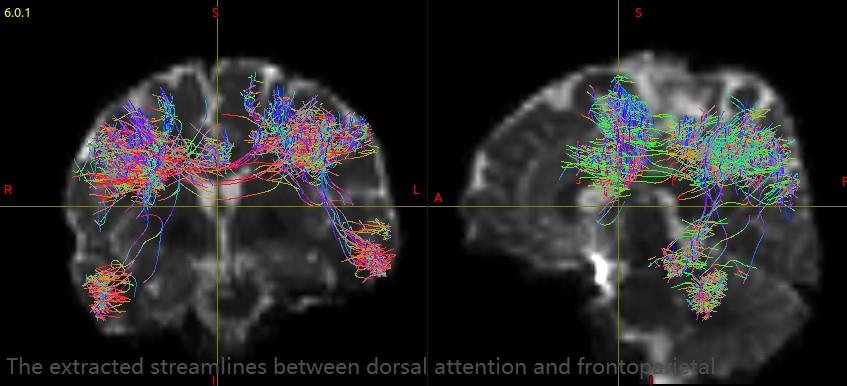
For HCP data, the GT results have a great improvement and very close to the results in your paper. I extracted streamlines between each pair of ROIs (Yeo’s 7 cortical modules plus subcortical) with connectome2tck. Almost all the extracted streamline bundles have lots of obviously spurious connectivity, such as the extracted streamlines between dorsal attention and frontoparietal. What could be the cause of these false streamlines? Is it the fiber tracking result or the altas I used? How can I improve this situation?
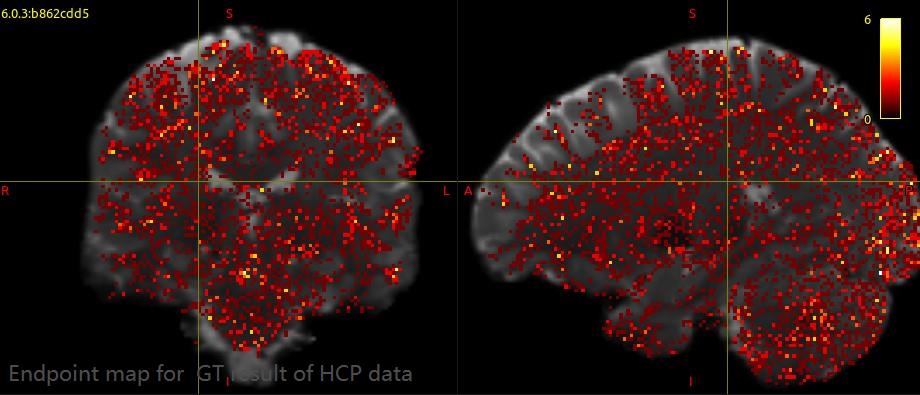
In addition, the streamlines endpoints of GT result are still distributed throughout the brain. I don’t know what the endpoint map should be look like for multi-shell multi-tissue GT. Did I get the right result? If not, how can I improve?
For single shell data, the GT results still have lots of short streamlines and didn’t have obviously improvement. The streamline bundles extracted from each pair ROIs include more short and spurious connectivity compared with HCP data, such as the extracted streamlines between dorsal attention and frontoparietal. How can I get better results? If this situation can’t improve, could you recommend any other global tractography algorithm for single shell data?
Glad to see that your results on the HCP data correspond with those in the paper. I don’t think those spurious fibres are unexpected – all tractography methods are sensitive to this. An exclusion region would suffice to take them out.
As I’ve said above, your single-shell b=1000 data is not well suited to the multi-shell multi-tissue model used in tckglobal so it’s not surprising that those results are subpar. If you are already using a 2-tissue model with CSF+WM responses and you have tried tuning -ppot and -cpot, there’s not much that can be done with this model and data I’m afraid.