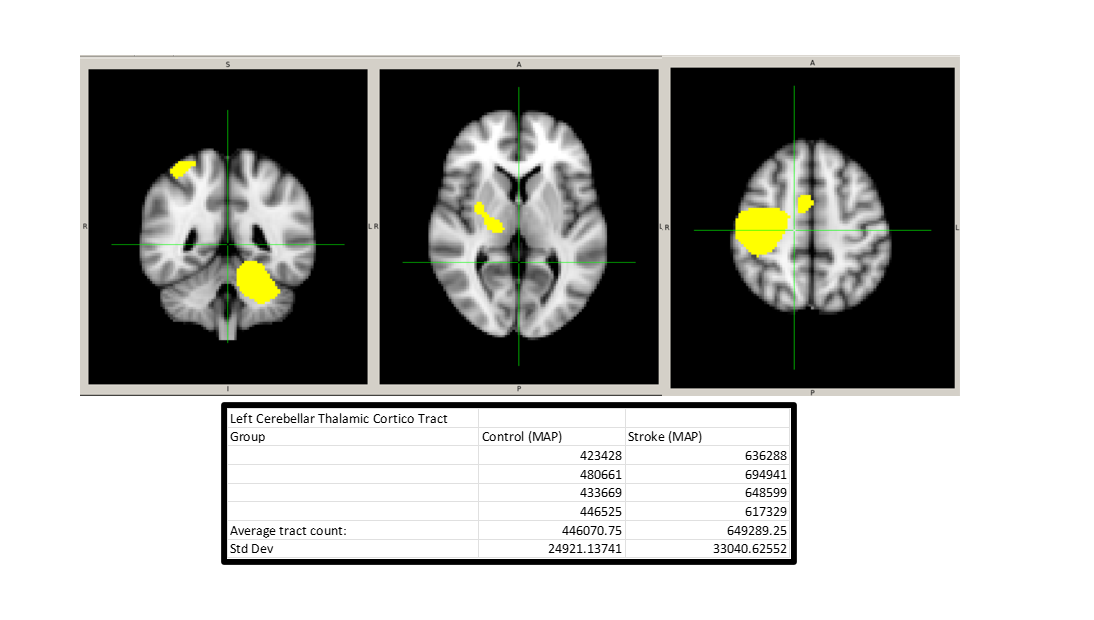
So currently I am extracting tracts between select ROIs. I employed a msmt_csd based ACT whole-brain tractography with selected 5m tracts between two cohort subsets (n=4 each). DWI acquisition was done through the same MAP sequence amongst participants. Preprocessing and tckgen were implemented with the same constraints across groups. When extracting the tracts within this designated network, which was determined via a group ICA in resting state fmri and co-registered to native diffusion space for each participant, there was an increase in tract count within the stroke group. CSF exclusion was implemented during the generation of the tractogram for both groups, assuming lesions as CSF. ACT tractography was also implemented in order to increase the anatomical accuracy of tracts.
Is this increase in tract count within the stroke group due to the restriction of pathways where reconstruction of said tracts can occur due to lesions (which increases CSF and exclusion zones).
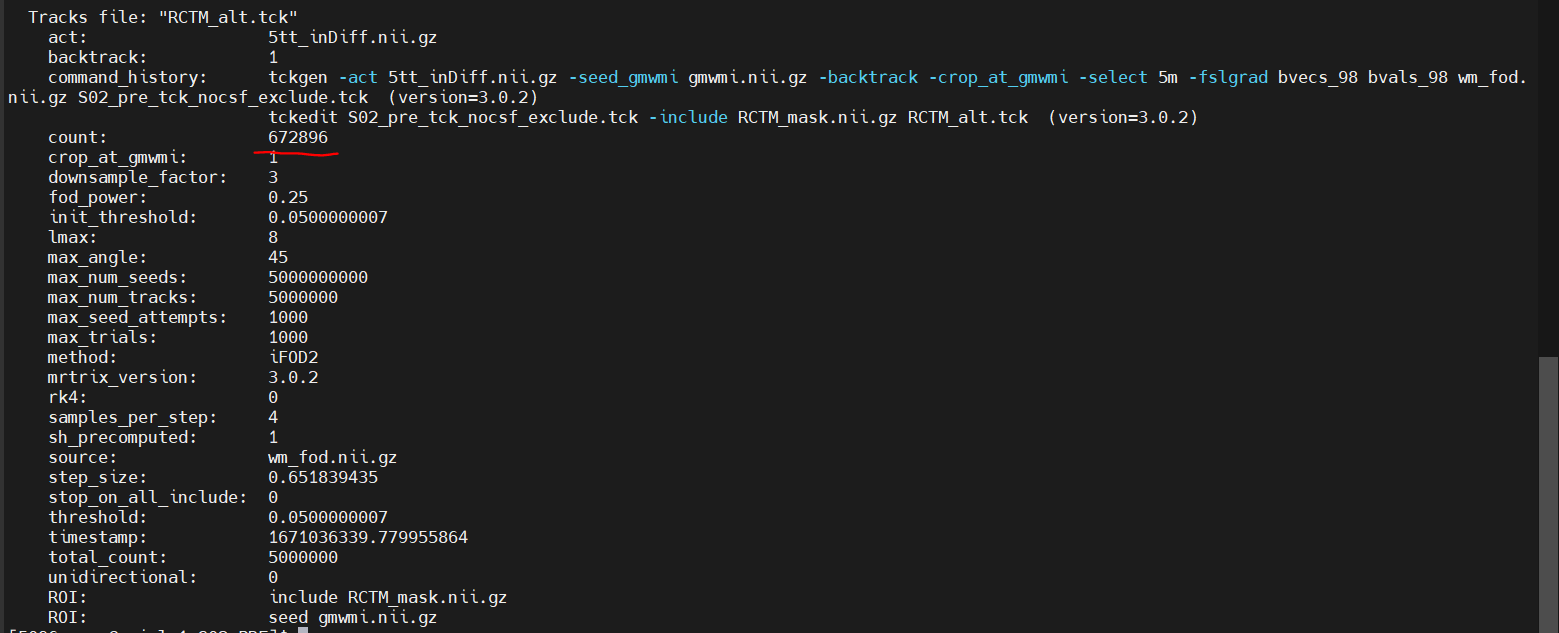
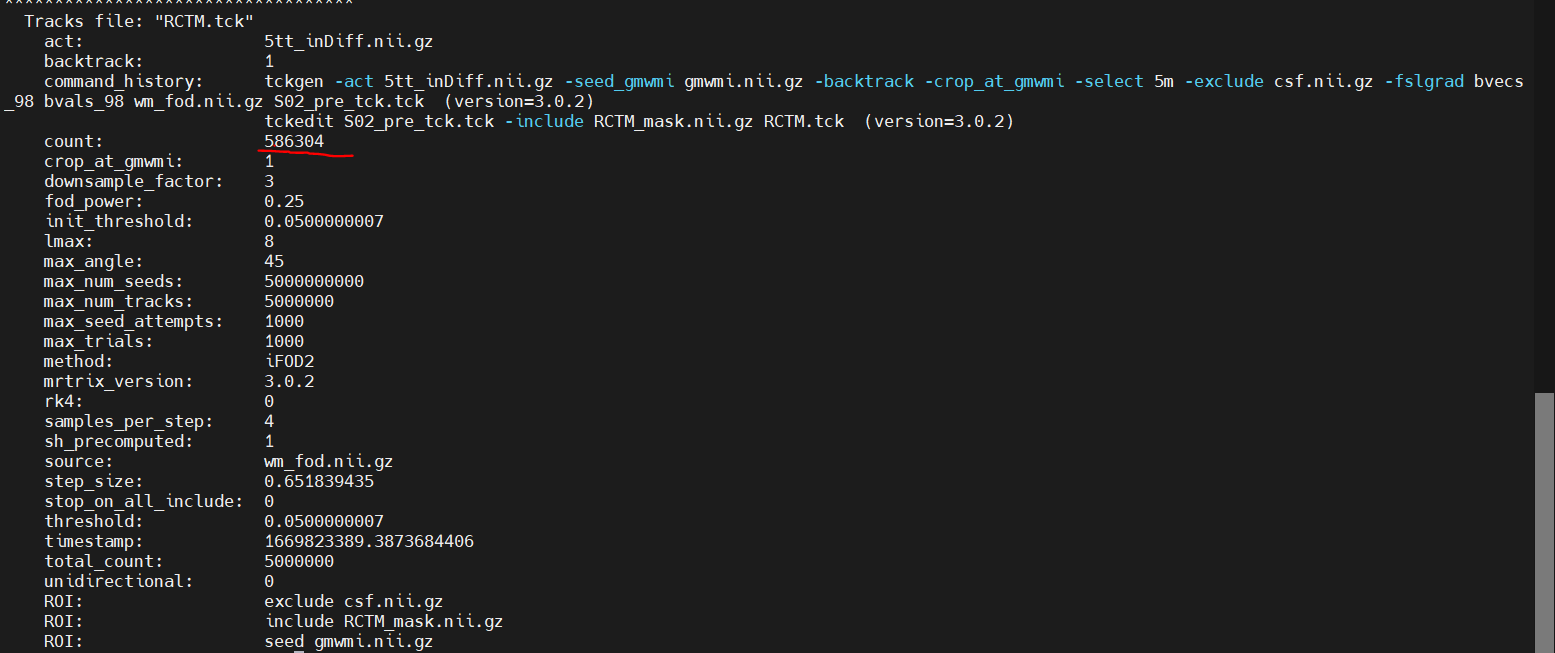
An update to this, I reran the tckgen without the exclusion of CSF and once again extracted the tracks within this network and it in fact did not help the situation, it actually increased the amount of tracks within the pathological case by quiet a bit within this subject (Added pictures of tckinfo describing the original construction of the whole brain connectome) and then the final counts of the subset extracted within the network.
Have you performed filtering of the tractogram after tractography? This may improve the comparability of the track counts (after tcksift) or track weights (after tcksift2) across your participants.
Could you make your inclusion mask (particularly the cortical and cerebellar regions) more localized? Right now they traverse large areas which might decrease the specificity of your resulting tracks. You could do this by creating an intersection of the functional ROI with the GM mask
Another way to increase specificity is to add the -ends_only flag to tckedit, so that only streamlines that end in the mask are included
Hey Nick! Thanks for the response. I like the ideas a lot. I’ve actually since posting this started to some of this already. I extracted endpoints within these rois and its still the case that there’s an increase in the stroke cohort. However, there a more asymmetric spread of endpoints, where more endpoints are present within the network affected by lesions is less than the one that is not (which is a good start).
I think making the ROIs of the network more focal is a grand idea that I’ll try later today. And I think this will definitely help improve this. The only issue is that because these areas are defined by an ICA, artificially making certain regions smaller may effect the overall interpretation. This is because these spatial spreads were determined to be p<0.05 across all subjects. But I am open to this and will try.
I was also thinking of doing an intersection mask between the ROI, and the gmwmi boundary obtained through the 5tt segmentation. Thoughts on this as opposed to doing a pure gm mask?
Again thanks for the thoughts, lets keep hashing this out. Best part of this process.
Glad that the patterns are looking more intuitive within your participants at least!
The functional data is low resolution and probably smoothed, so in any case you can’t be certain that the activity is completely localized to those voxels. Given the higher chance of false positives (low specificity) in the tractogram, it might be worth trying to limit the spatial extent of the network to improve the specificity of your findings. You could still keep your ICA network structure and use a higher threshold (for example p<0.001) - whichever network detection threshold you use is somewhat of an arbitrary choice
Either the cortical ribbon (might be easier to interpret when you view it) or gmwmi should be fine.