Hi Mrtrix users,
Im Takafumi Yano. Nice to meet you!
In our labs, single shell diffusion MR data are used (32 gradients b=1000 and b0).
And, we want use mrtrix for the tractography method.
Then, we tried to use tckglobal command like this.
dwi2response msmt_5tt dwi.mif 5tt.mif wm.txt gm.txt csf.txt
tckglobal dwi.mif wm.txt -riso csf.txt -riso gm.txt -mask mask.mif -niter 1e8 -fod fod.mif -fiso fiso.mif tracks2.tck
Unfortunatley, the result is not that we assumed.
Plese, suggest me any idea to overcome this problems… 
Thank you!
Takafumi Yano
That’s going to be quite challenging… Other people with low b-value single-shell data have reported some success by adding a CSF compartment; but I would highly advise to leave out the GM one. See this post (link) for a long description of options I gave to a few other users, and some results posted by a user further on in that thread (scroll up to get more context, scroll down for results).
In the interest of getting a more accurate CSF response function, I would advise to use the dhollander algorithm in dwi2response as well; we’ve often found that (apart from being more convenient to use; no 5tt segmentation and registration of T1w image to diffusion data required) specifically the CSF response function from the msmt_5tt algorithm can be (quite) suboptimal. The dhollander algorithm doesn’t seem to have this problem.
Using the instructions in the aforementioned post, you could go for dwi2response dhollander followed by dwi2fod msmt_csd, but the latter one with only the WM and CSF responses (ditching the GM one at this stage); and then followed by tckgen for probabilistic streamline tractography (which will allow you to use the ACT framework for anatomical constraints as well if you provide it with your 5tt segmentation; this should particularly benefit you well at a low b-value and in the absence of a GM compartment).
If you want to go for tckglobal, it may be worth a shot trying this as well without the GM response (so again just the WM and CSF one)… but I’m not sure how well that would work for such limited data (low b-value and limited number or gradient directions). @dchristiaens can probably comment further on this option.
Hi, Thijs,
Thank you for your reply and very detailed suggestion.
I appreciate your quick response and kindness.
I tried these your suggestions.
I used dwi2response dhollander scripts and tckglobal.
$ dwi2response dhollander dwi.mif out_sfwm.txt out_gm.txt out_csf.txt
$ tckglobal dwi.mif out_sfwm.txt -riso out_csf.txt -mask mask.mif -niter 1e8 -fod fod3.mif -fiso fiso3.mif tracks3.tck
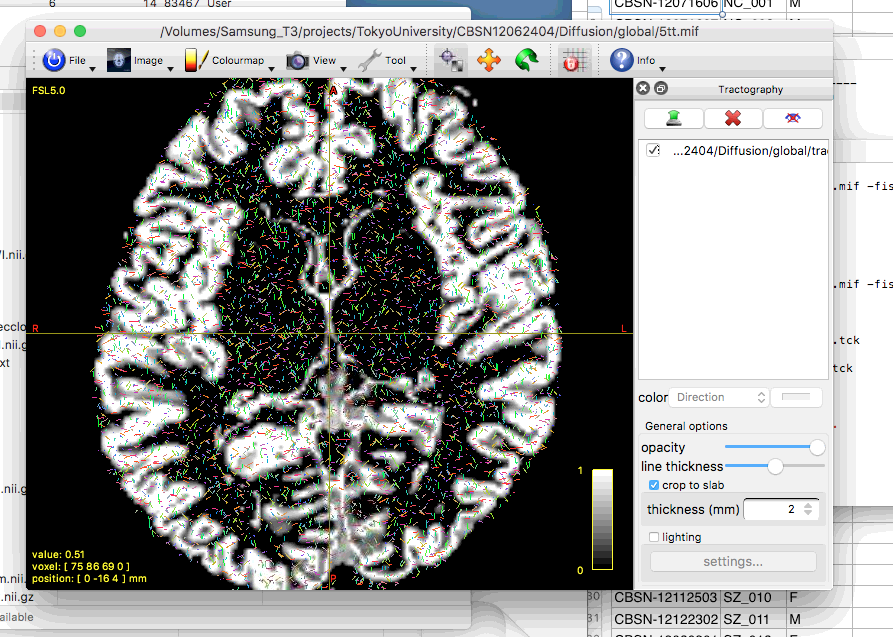
But unfortunately, I could not do global tractography correctly.
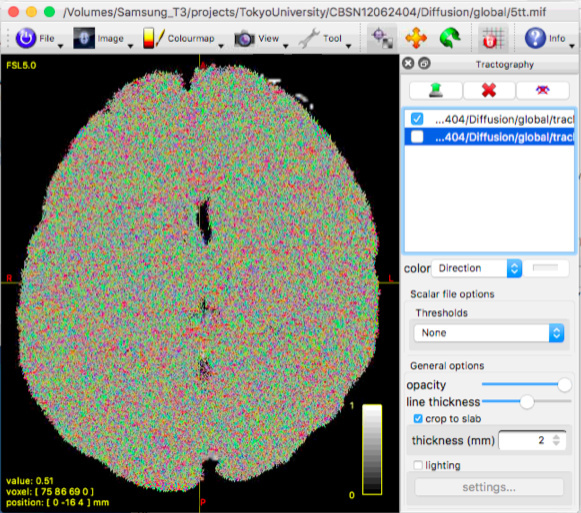
This figure represents the result with WM and CSF response function.
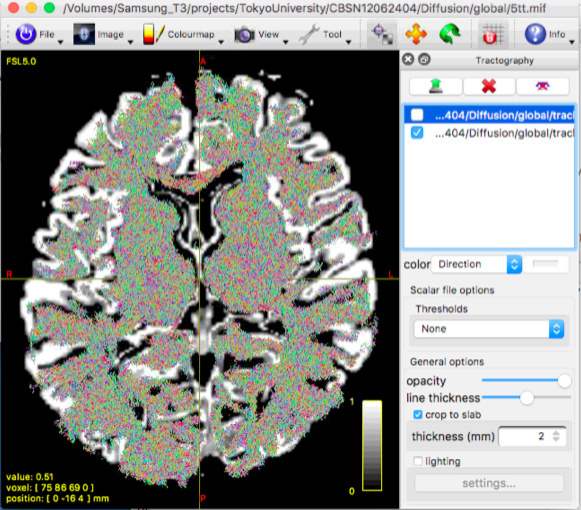
This figure represents the result with WM, GM and CSF response function.
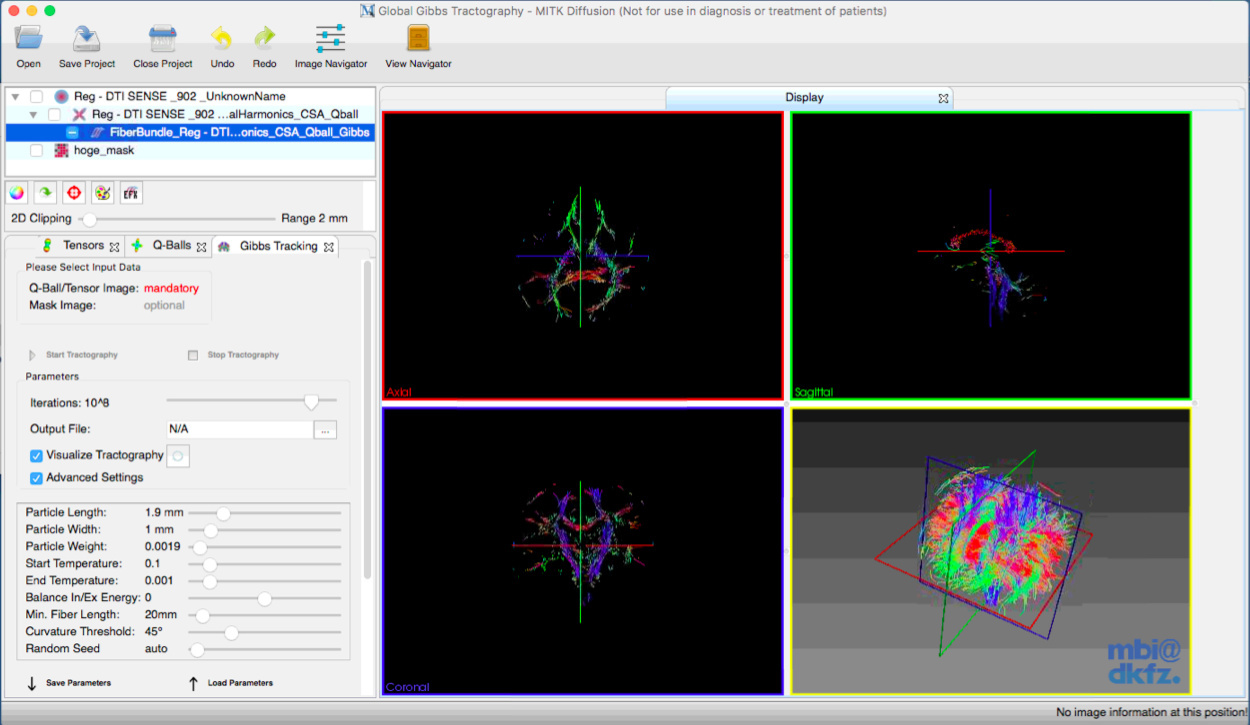
And, I further used MITK global tractography.
This is a result from MITK diffusion global tractography.
I think MITK diffusion’s global tractography can estimate properly.
But, MITK’s global tractography method was developed earlier than mrtrix of it.
Then, I simply think that mrtrix also could estimate tractography with low b and a small number of gradient directions.
I wrote the procedure in MITK global tractography.
- import DICOM files.
- bet (fsl command) and estimate the brain mask.
- Q-ball imaging with solid angle method.
- Gibbs global tractography
Is there any way to do that with mrtrix.
And, if you know why MITK method do that but, mrtrix couldn’t do that, please tell me.
Thank you,
Takafumi Yano.
Dear Takafumi,
I would also recommend using a WM-CSF model for single shell data, regardless of which heuristic you use to obtain the response functions. Do check that your WM single-fibre response has the expected pancake-shape to be sure.
In order to make tckglobal work for your data, you will have to tune the algorithm parameters a bit better. The defaults really assume a more complete multi-tissue model and multi-shell HARDI data. Just eyeballing your latest screenshots, I think you will likely need to increase the particle potential (e.g. -ppot 0.1) and/or its weight (e.g. -weight 0.2). Depending on the voxel size of the DWI, you may also want to increase the segment length (e.g. -length 1.5 or 2.0).
In order to test and compare different parameter settings more easily, you can draw a mask for any smaller region of interest. I usually to take a cube of about 10x10x10 voxels around the (left or right) semioval centre. Then, you can run tckglobal within that block for a smaller no. iterations (e.g. 1e7). When you’re happy with the parameter settings, apply them in the full brain mask but scale the no. iterations with the volume ratio between both (to about 1e9) in one long run.
MITK global tractography of course uses a different model (single-shell ball-and-stick) whereas our method was designed to leverage multi-shell data with a multi-tissue spherical convolution model, but I don’t expect that to have such a big impact on the fibre reconstruction in your data. A more likely cause relates to the issue of parameter selection I discussed above. MITK has implemented a clever way of automatically tuning some of the parameters behind the scenes at the start of the MCMC process, using heuristic criteria of the energy evolution during the burn-in phase. This means that some of the parameters in the “advanced settings” panel can differ between protocols and between datasets. Unfortunately, we do not have such automated calibration procedure in place in tckglobal. In my experience, the defaults work fairly well for most multi-shell HARDI data, but different protocols such as yours require a bit of manual tuning. I hope that the guidelines above will be of help. If not, it’s probably easier if you email me a dataset, so I can have a look.
Cheers,
Daan
Dear Dchristiaens,
Thank you for your reply.
I tried to change some parameter.
Unfortunatlely, the result is not we assumed.
The changes is below
-weight 0.1 to 0.001 and 0.2
-ppot 0.1
-iteration 1e6 to 5e7
Then, I send you my data. If you can please check my data.
I think MRTRIX could be applied to low b data, because MITK’s method can do it…
Thank you,
Takafumi Yano.