Hi all,
I am creating Structural Connectome Matrix for a preterm neonatal brain (Gestational Age=32 weeks).
I wanted to make sure the steps I am adopting are correct.
My data consist in:
- dki_bfc.mif → diffusion image after preprocessing with Mrtrix3 (denoising, unringing, distortion corrections and bias field corrections) (resolution: 1.55556 x 1.55556 x 2.2 x 1)
- 3dT1_corr_brain.nii.gz → structural T1 image after bias field correction and skull stripping (resolution: 0.890003 x 0.885417 x 0.885417)
- mean_b0_BRAIN_corr.nii.gz → mask image in diffusion space
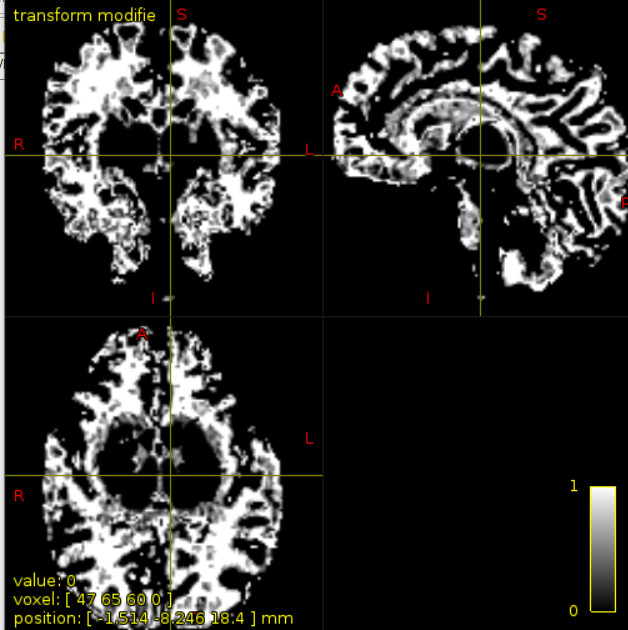
First of all, I registered skull-stripped T1 image to mean b=0 diffusion image, though keeping its higher resolution through trasformconvert command
flirt -in mean_b0_BRAIN_corr.nii.gz -ref 3dT1_corr_brain.nii.gz -dof 6 -omat DKI_2_T1.mat
transformconvert DKI_2_T1.mat mean_b0_BRAIN_corr.nii.gz 3dT1_corr_brain.nii.gz flirt_import DKI_2_T1_mrtrix.txt
mrtransform -linear DKI_2_T1_mrtrix.txt -inverse 3dT1_corr_brain.nii.gz ${SBJDIR}/T1_in_DKI.nii.gz
Then, I computed 5TT Nifti for ACT/SIFT using -premasked option since T1 is already skull stripped
mrconvert T1_in_DKI.nii.gz anat.nii
5ttgen fsl anat.nii 5tt.mif -premasked
I computed GMWMI Nifti for ACT/seeding
5tt2gmwmi 5tt.mif gmwmi.mif
I estimated msmt response function
dwi2response msmt_5tt dki_bfc.mif 5tt.mif resp_wm.txt resp_gm.txt
I computed msmt FODs
dwi2fod msmt_csd dki_bfc.mif -mask mean_b0_BRAIN_corr.nii.gz resp_wm.txt fod_WM.mif resp_gm.txt fod_GM.mif resp_csf.txt fod_CSF.mif
I downsample FODs for faster tracking using mrgrid
mrgrid fod_WM.mif regrid -scale 0.5 -interp sinc fod_WM_ds.mif
I generated first tractogram (50million streamlines)
tckgen fod_WM_ds.mif tracks_msmt_50M.tck -act 5tt.mif -backtrack -crop_at_gmwmi -seed_gmwmi gmwmi.mif -maxlength 250 -select 50M -cutoff 0.01 -step 1
I applied SIFT to reduce to 5million streamlines
tcksift tracks_msmt_50M.tck fod_WM_ds.mif tracks_msmt_5M_01.tck -act 5tt.mif -term_number 5000000
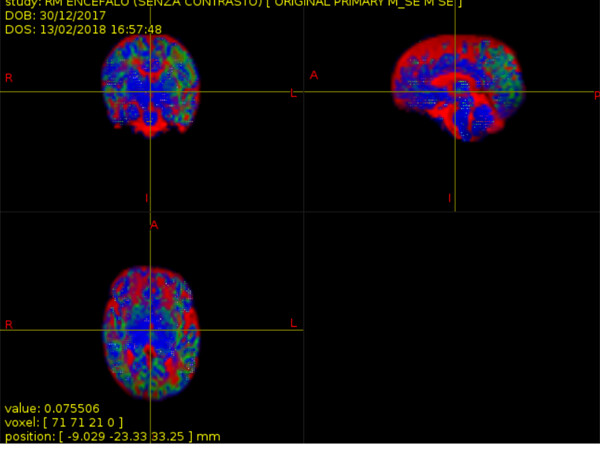
As atlas, I wanted to use skull-stripped T1 of JHU neonate atlas (https://cmrm.med.jhmi.edu/cmrm/Data_neonate_atlas/atlas_neonate.htm). It has 122 parcellations listed from 1 to 122 in the attached file (JHU_neonate_lut.pdf) , and interleaved from right and left hemisphere. I registered those ROIs (LABELS_dki_corr.nii.gz) to diffusion space using nearest neighbour interpolations.
My question now concerns how to generate the connectome matrix from this starting point.
Since, Look-Up table is already well-ordered I thought of skipping labelconvert command, and simply run:
tck2connectome tracks_msmt_5M_01.tck LABELS_dki_corr.nii.gz connectome.csv
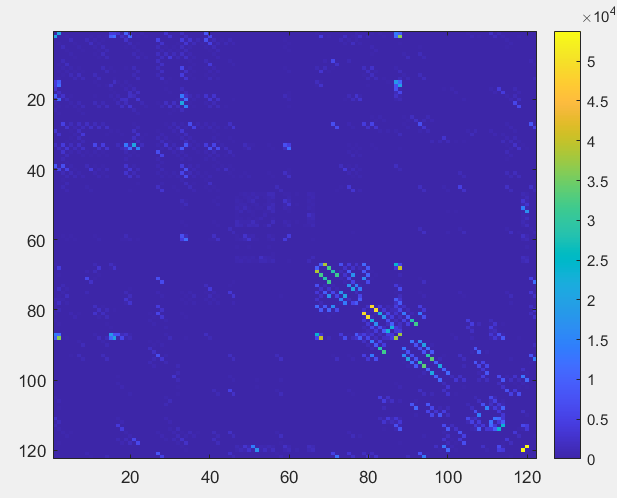
Is this correct or should I organize and select parcels? Can I use all of them (122)?
Doing so the resulting connectome visualization results as follows, after importing it in MATLAB
sc=importdata('connectome.csv')
sc=triu(dt,1)+triu(dt,1)'
imagesc(sc)
 Processing: JHU_neonate_lut.txt…
Processing: JHU_neonate_lut.txt…
JHU_neonate_lut.pdf (34.9 KB)