Hello, I really appreciate the amount of help available on this forum, thank you!
I just started using mrtrix3 and was hoping someone could give feedback on below processing steps and dwi2fod output
The rawdata (dwi_raw.mif) has resolution 1x1x2, 66 volumes, with 10 b0s and 56-dirs with b=1000;
below is what I have done so far-
-
dwidenoise dwi_raw.mif dwi_deno.mif -noise noise.mif
-
mrdegibbs dwi_deno.mif dwi_deno_unr.mif -axes 0,1
-
dwifslpreproc dwi_deno_unr.mif dwi_den_unr_preproc.mif —nocleanup rpe_none -pe_dir AP -eddy_options " --slm=linear" -fslgrad bvec bval
-
dwibiascorrect ants dwi_den_unr_preproc.mif dwi_den_unr_preproc_unbiased.mif -bias bias.mif
5a) mrconvert dwi_den_unr_preproc_unbiased.mif dwi_den_unr_preproc_unbiased.nii
b) bet2 dwi_den_unr_preproc_unbiased.nii dwi_den_unr_preproc_unbiased_masked.nii -m -f 0.7
c) mrconvert dwi_den_unr_preproc_unbiased_masked.nii mask.mif
-
dwi2response tournier dwi_den_unr_preproc_unbiased.mif wm_response_tournier.txt
-
dwi2fod csd dwi_den_unr_preproc_unbiased.mif wm_response_tournier.txt fod.mif -mask mask
The output looks like below-
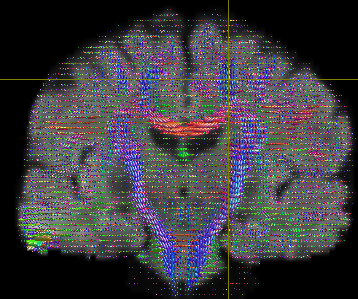
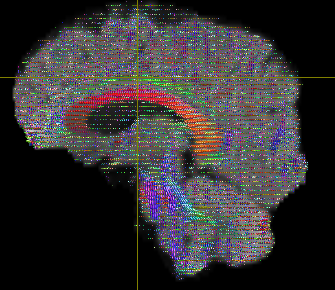
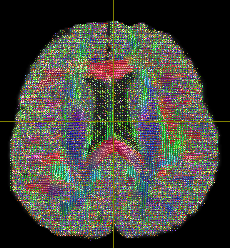
First question is if I should modify any steps from 1-7 to improve this output?
Secondly my goal is do prob tractography and compute connectogram using a white matter atlas; is there a webpage that details the steps? I found the https://osf.io/pm9ba/ page and https://andysbrainbook.readthedocs.io/en/latest/MRtrix/MRtrix_Introduction.html, but the former is for connectome multi-shell data, and the latter explains with freesurfer outputs; I would like to try an atlas based method.
Thanks so much!