I’ve been trying out various combinations of tckgen and tcksift options on our dataset and so far I haven’t found a satisfactory combination. I was wondering I might be missing something or if there was some kind of general guidance available for this situation.
The dataset: 1.8x1.8x2.5mm, 64 directions in a single b=800 shell (+ 1 b0), and includes a standard EPI fieldmap as well as a 1mm T1 anatomical image. We performed basic topup and eddy correction, though I have noticed some residual disagreement between diffusion and T1 around the frontal pole for many subjects. We have run freesurfer on the T1, and performed manual editing of these freesurfer reconstructions. Many of our subjects have white matter abnormalities, so we would prefer to use these manually edited segmentations where possible. T1 was registered to diffusion after freesurfer.
We’ve been reading about the reduced false positive rates using CSD+deterministic tractography+filtering, and based on this post on single shell data, our current rough-draft pipeline is:
dwi2response dhollander DWI.mif RF_wm_dhollander.txt RF_gm_dhollander.txt RF_csf_dhollander.txt -voxels RF_voxels_dhollander.mif
dwi2fod msmt_csd DWI.mif RF_wm_dhollander.txt RF_wm_dhollander.mif RF_csf_dhollander.txt RF_csf_dhollander.mif
tckgen RF_wm_dhollander.mif CSD_sdstream_1M.tck -algorithm SD_STREAM -seed_dynamic RF_wm_dhollander.mif -select 1M
tcksift CSD_sdstream_1M.tck RF_wm_dhollander.mif CSD_sdstream_1M_sift500K.tck -term_number 500k
I realize 1M->500K is not a very rigorous filter, and we could use larger values, but I’m hesitant to commit significant compute resources (we have >100 subjects) until I see outputs that suggest I’m on the right path. The current outputs are dominated by short fibers within gray matter 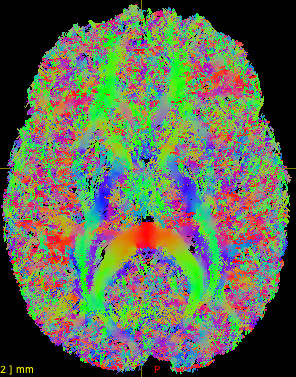 . I would like to restrict these in a meaningful way without being overly prescriptive. I have played with running
. I would like to restrict these in a meaningful way without being overly prescriptive. I have played with running 5ttgen freesurfer (to make use of our manual edits) and using the -act options in tckgen and tcksift. Just running tckgen with -act has taken tractography from 40 minutes to what looks like 6 or more hours per subject, not including tcksift.
As I said, if this is the “correct” approach, I’m willing to go for it, but I was hoping for some expert input before doing so, especially since there are many additional algorithm or parameter choices that I haven’t even considered that might be having more of an impact in this case.
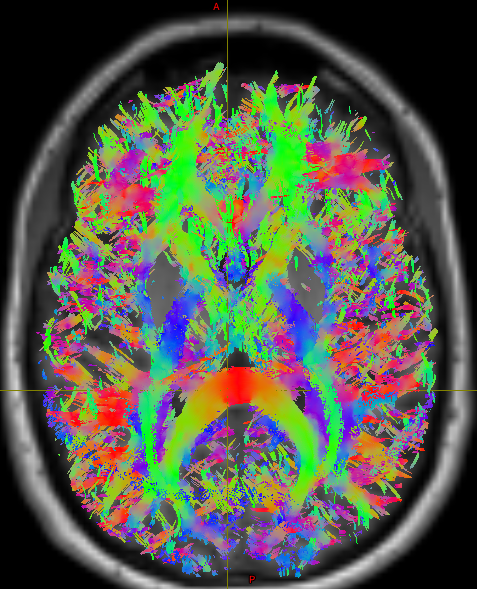
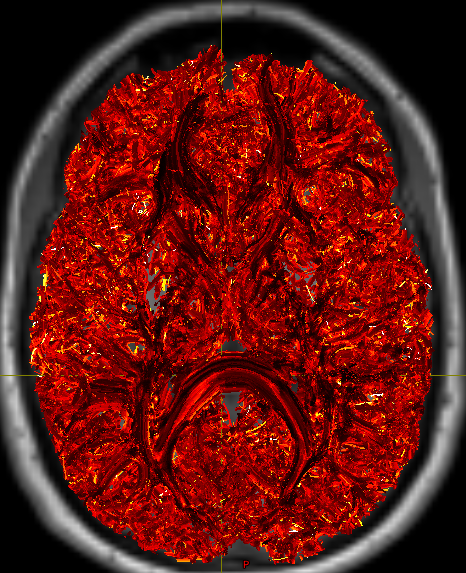
 Good to hear this conversation was helpful. Thanks
Good to hear this conversation was helpful. Thanks  The one in
The one in 