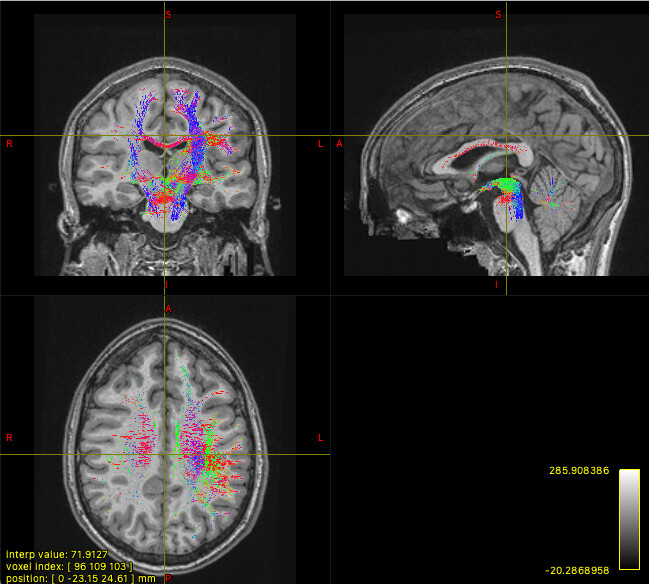
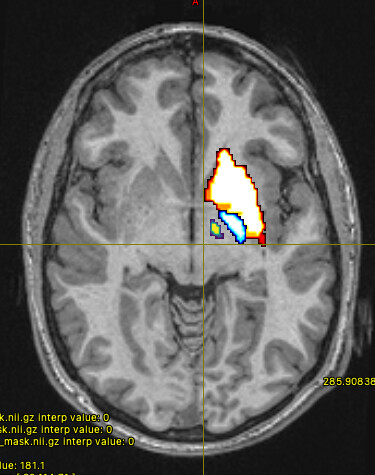
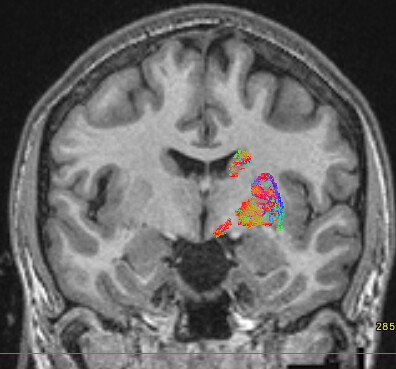
The mask option isn’t really suitable in this context, since it will simply truncate any streamlines that exit the mask. The streamlines you’re seeing may well have connected between your ROIs, but the use of -mask will clip off the parts outside of the mask, does that make sense? So you could create a mask encompassing all of your ROIs, but that doesn’t mean that the streamlines you’ll see originally remained within that mask, you’re just hiding the bits that didn’t!
The streamlines selected using -include only have to pass through your regions of interest at a minimum. As long as a streamline touches each of your masks, it’s free to go wherever else it pleases!
So your initial results are what you would expect, since the sreamlines passing through your ROIs are likely to visit other parts of the brain. (As Marcos points out, in probabilistic tractography, most generated streamlines are probably going to be “wrong” one way or another.)
If you only want to see streamlines that terminate in a specific ROI you could try the -ends_only flag, but I’m not sure how you would make that work with 3 ROIs, when a streamline only has 2 ends! You’d probably have to do the different combinations separately e.g.
tckedit wbt.tck -include A.nii.gz -include B.nii.gz -ends_only A_to_B.tck
tckedit wbt.tck -include B.nii.gz -include C.nii.gz -ends_only B_to_C.tck
# etc
You could also try specifying a -maxlength to narrow your results down to shorter streamlines local to your ROIs.
Ultimately you’ll be limited by whatever streamlines are already present in your whole-brain tractography, so the parameters used to generate those may or may not have been optimal for seeing the connections you’re interested in. Short streamlines in particular around that area are unlikely to be abundant - in particular any streamline passing through your dorsal striatum ROI is on a fast track to the entire frontal lobe! Again, specifying a -maxlength at this stage might help, but would be computationally pretty wasteful.*
I would be careful about about constraining and wrestling your streamlines too much though, otherwise you risk your results becoming uninterpretable. To paraphrase a great scholar: “If you torture your tractography algorithm, it’ll tell you anything!”
*if my interpretation of -maxlength is correct, that it discards any tracks that exceed the limit, rather than truncating them.