Hi!
I have sift2 processed whole brain 2M connectomes, with the corresponding separate weights file.
I have been able to load both to matlab, threshold the fibers and write the tck file back, but it does not seem like a good method.
Is there a way within mrtrix to do this?
Is there any recommendation on how to choose the threshold to delete fibers?
Thanks!
Gari
tckedit with the -minweight option should be designed exactly for that.
Depends probably on why you would want to threshold individual streamlines based on their weight to begin with… I’m not sure if there’s even a good reason to do so in any scenario. @rsmith may be able to help you further, but it would probably help if you give a bit more explanation on what your (study/experiment) goal is exactly.
Thanks for your answer,
I am using the mrtrix pipeline in conjunction with other analysis pipeline.
I already had some tracts identified with a tool, but based on a whole brain fiber tractography calculated with another tool.
Now I would like to identify the same tracts using the same identification program, but base the tract identification on the whole brain fiber tractography calculated with mrTrix.
This program cannot use the weights information coming from mrTrix.
I assume (maybe wrongly, please correct  ), that if I obtain the whole brain fiber tractography, and correct it with sift2, it would be recommendable to clean the fibers with low weights before using the tract identification program. Once I do the cleaning, all fibers will have a weight of 1, so to speak.
), that if I obtain the whole brain fiber tractography, and correct it with sift2, it would be recommendable to clean the fibers with low weights before using the tract identification program. Once I do the cleaning, all fibers will have a weight of 1, so to speak.
What do you think?
Thanks!
Gari
Hi Gari,
Quite a number of things to cover of here.
Is there a way within mrtrix to do this?
-
tcksift2includes options-min_factorand-min_coeff, which will remove any streamlines from the tractogram if their streamline weighting factor / coefficient fall below a specified threshold. This has the benefit that the model will continue to be refined given the perturbation introduced by the removal of that streamline. -
tckeditdoes indeed have the-minweightoption for this purpose. However, on a conceptual level: If you globally optimise some model, then remove bits and pieces of that model, the model is no longer optimal. Those low-weight streamlines were used as part of the optimisation; their removal may result in there being insufficient streamlines density in those fixels they traverse, which would ideally be offset by a slight increase in the weights of other streamlines traversing those fixels. While this effect may be small if the weights of the removed streamlines are small, it is nevertheless a bad approach to adopt.
Is there any recommendation on how to choose the threshold to delete fibers?
Assuming you feel the urge to remove streamlines (which I find to be misplaced more often than not), I would suggest taking a closer look at Figure 8 in the manuscript. However for the most part: Streamlines with little weight have little contribution to either the model, or any result being quantified; their removal requires an arbitrary threshold, which unnecessarily increases the complexity of your processing chain; low-weight streamlines are not strictly false positive streamlines (in fact if a geometrically-simple, large-density pathway is easy to track, and hence contains many streamlines, those streamlines will have low weight, so it can in fact be the opposite).
This program cannot use the weights information coming from mrTrix.
If whatever other software you are using is unable to properly use / interpret the weights from SIFT2, yet your analysis / quantification is wholly dependent on the SIFT model being accurately fit, then I would suggest that you probably should be using SIFT rather than SIFT2.
I assume (maybe wrongly, please correct
), that if I obtain the whole brain fiber tractography, and correct it with sift2, it would be recommendable to clean the fibers with low weights before using the tract identification program.
Similar to before. I personally advise against such “cleaning”, and am not aware of any articles where such removal has been performed. Again, it’s likely a false equivalence between “low weight” and “false positive”. The SIFT model does not have any capability to distinguish between these two; or if it does, it’s not beating chance by very much.
Once I do the cleaning, all fibers will have a weight of 1, so to speak.
Most definitely not. Again referring to Figure 8 in the manuscript, the weights within the tractogram have a distribution, and it is the optimisation of the tractogram with respect to the image data that yields that distribution. If you were to truncate that distribution such that streamlines with a weight below some threshold are instead removed from the reconstruction, that may not influence the model overall a great deal; but if you were to take all streamlines surviving that threshold and fix all of their weights back to 1.0, this would completely destroy the optimisation performed by SIFT2, the tractogram wouuld no longer provide a reconstruction of the underlying fibre density estimates, and the accuracy & interpretation benefits provided by the SIFT model optimisation would no longer be applicable to your data. This again advocates the use of the SIFT algorithm rather than SIFT2 in your use case.
Cheers
Rob
1 Like
Thanks for your detailed answer Rob!
Yes, I think in this precise use case I should use sift instead of sift2.
As far as I remember, should I run the tracking again with an order of magnitude higher number of fibers in order to make sift work better? (so, 500K >> 5M)
Thanks again!
Gari
PS When I said weight 1, I meant that the other program would not differentiate depending on weights, it would consider it or not for the fascicle creation and evaluation, 0 or 1.
… should I run the tracking again with an order of magnitude higher number of fibers in order to make sift work better?
Yes, it’s of that order of magnitude. If e.g. two voxels in the brain contain the same underlying fibre density, but one contains 10 times as many tracks as the other, you need to remove ~ 90% of the tracks in order to correct that reconstruction bias. That’s a very simplified explanation, which doesn’t take into account the fact that each streamline traverses many voxels and hence performing the correction is more complex. Removing 90% of streamlines has been used fairly commonly, so you can blame somebody else for that decision ![]()
PS When I said weight 1, I meant that the other program would not differentiate depending on weights, it would consider it or not for the fascicle creation and evaluation, 0 or 1.
Yes, I understood your intention. What I meant is that those weights are the principal output of SIFT2, and what it uses to provide quantitative properties to the tractogram; therefore, if you take the results of SIFT2, and provide them to some other software that is not capable of interpreting the weights of SIFT2, then that is equivalent to never having run SIFT2 in the first place. Selectively removing only streamlines to which SIFT2 assigns very small weights, then effectively resetting the weights of all retained streamlines to 1, would be comparable to running the SIFT algorithm, with that number of streamlines having been removed.
Hi Rob
I have a similar question regarding thresholding connectomes generated with SIFT2 (and ACT+freesurfer). Here is my average connectome across young subjects (I have a group of old subjects as well). Note I also multiplied them with mu which resulted in values between 0-~2.5 with some values in order of e-4. I hope these small numbers do not have any effect on network analysis further down the road.
I guess the overall pattern of my connectomes fits well with those in Yeh et al., 2015 (knowing you used SIFT there! ![]() ). right?
). right?
Original:
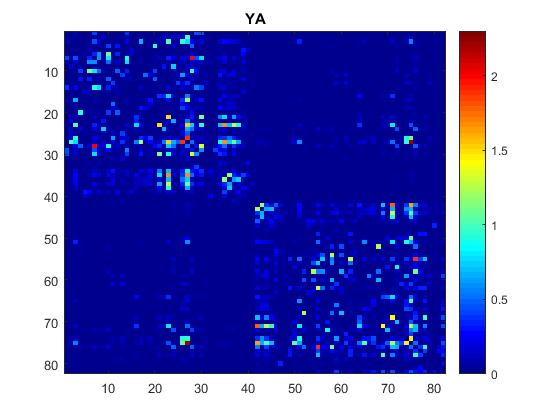
and after playing a bit with the values to better visualize:
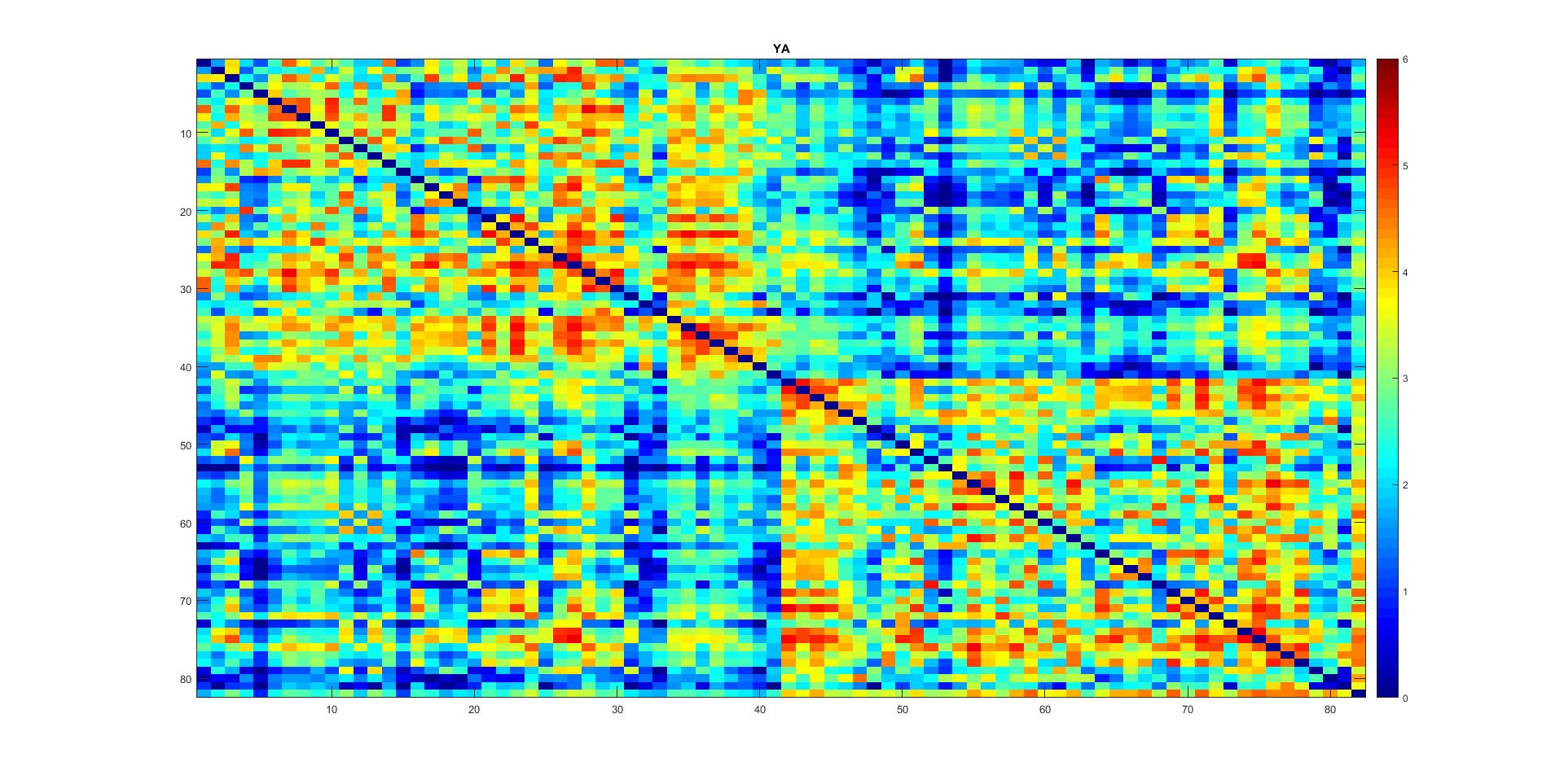
I see that in many connectomic papers, the generated connectomes are thresholded.
So as far as I understood from your explanation above, it is better not to threshold the SIFT2 connectomes. right?
I am trying to use GraphVar software which incorporates Brain connectivity toolbox functions. Could you please comment on these:
I should use the weighted undirected version of all network metrics.
There are a lot of random networks to use. Which one do you suggest to use in this case?
My average weighted degree is 79. Is this normal? Does this mean that the SIFT2 connectomes are almost fully connected?
Thanks and cheers,
Hamed
Hi @Hamed,
My viewpoints to some of the questions you raised:
Ideally, we would not want to use a threshold; yet, more experiments are needed to understand the practical influences (e.g. on network analysis). My colleague is currently looking into this, and hopefully there will soon be some conclusions to your question.
Yes, I believe that a weighted version should be more meaningful since it tends to capture the connection heterogeneity of biological fibres, which is adversely completely lost in an unweighted (binary) method.
An undirected version should be used since tractograms do not provide the information transfer of neural signal.
I used BCT’s function for generating undirected random graphs; haven’t tried other randomization methods.
The degree metric is not quite informative in this case since it is very sensitive to a number of factors in the process of connectome construction. Also, my personally opinion is that many weighted network metrics that are degree-dependent need to be revisited for tractogram-based connectome analyses (as described in the article you mentioned).
Cheers.
Hi Rob @rsmith !
I have tried to use these two methods to reduce the number but failed. I’m not sure I understand this guideline of needing to delete 90% tracts. Could you please help me to check if the following three approaches are appropriate or not?
tcksift2 -min_factor 0.9 ...
# no further tckedit is needed
tcksift2 ...
tckedit -tck_weights_in weight.txt -minweight 0.9 ...
tcksift2 ...
tckedit -number "$number_tckgen/10" ...
Thank you and cheers,
Ke