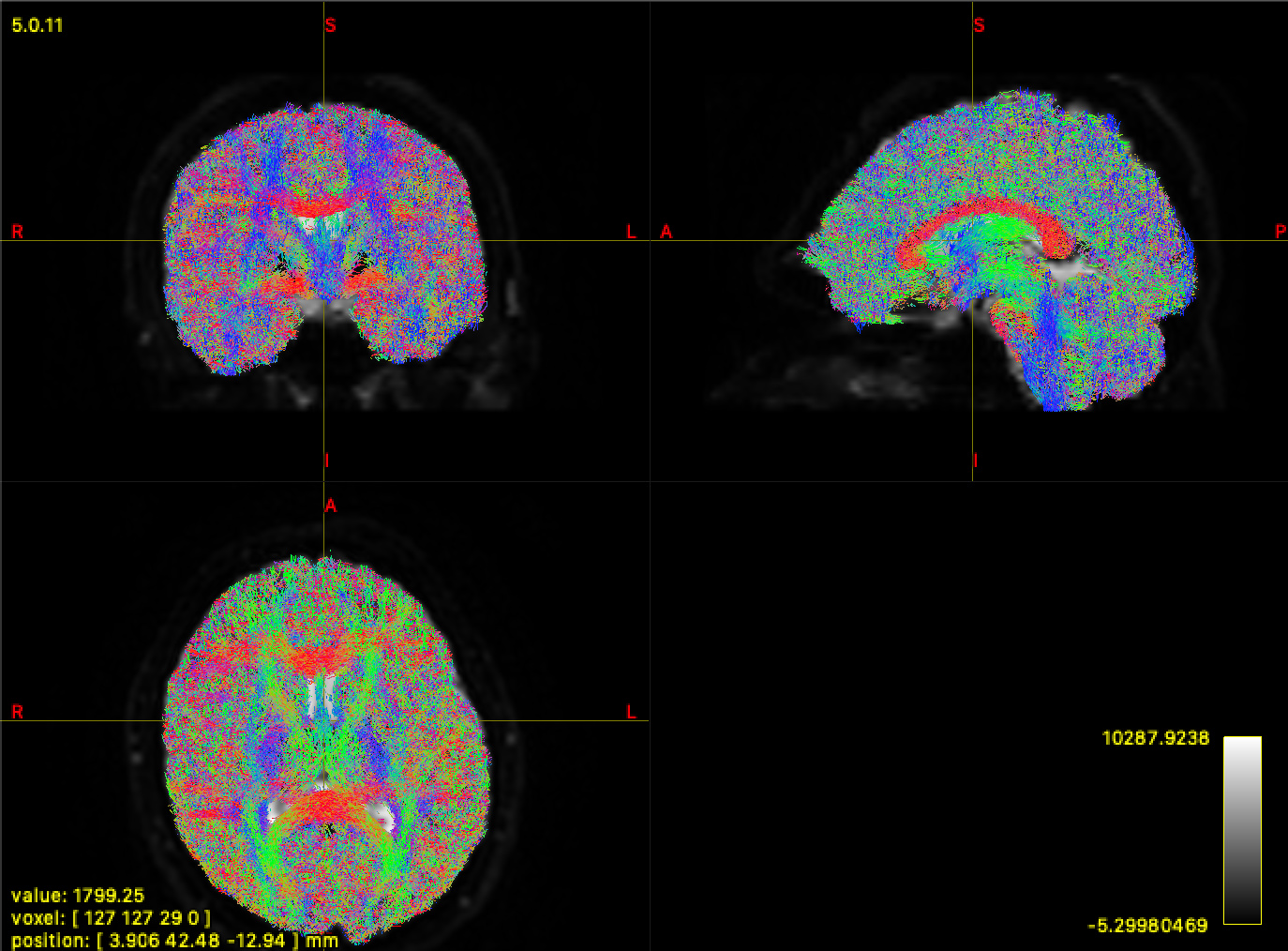
I have single shell DWI data with b=0 & b=1000, with no distortion correction unfortunately, so I have not been using ACT. I’ve run through the entire pipeline and I get the result below for my tractography. I shouldn’t expect so many streamlines in the grey matter.
From reading the docs and many forum posts, I ran the dhollander algorithm to get the tissue response functions and then only calculate the ODF for WM and CSF because of single shell data:
However, this gives me a FOD_WM that looks weird for the b0 volume. I’m not entirely sure what to expect here, but my suspicion is this may be throwing off the tractography.
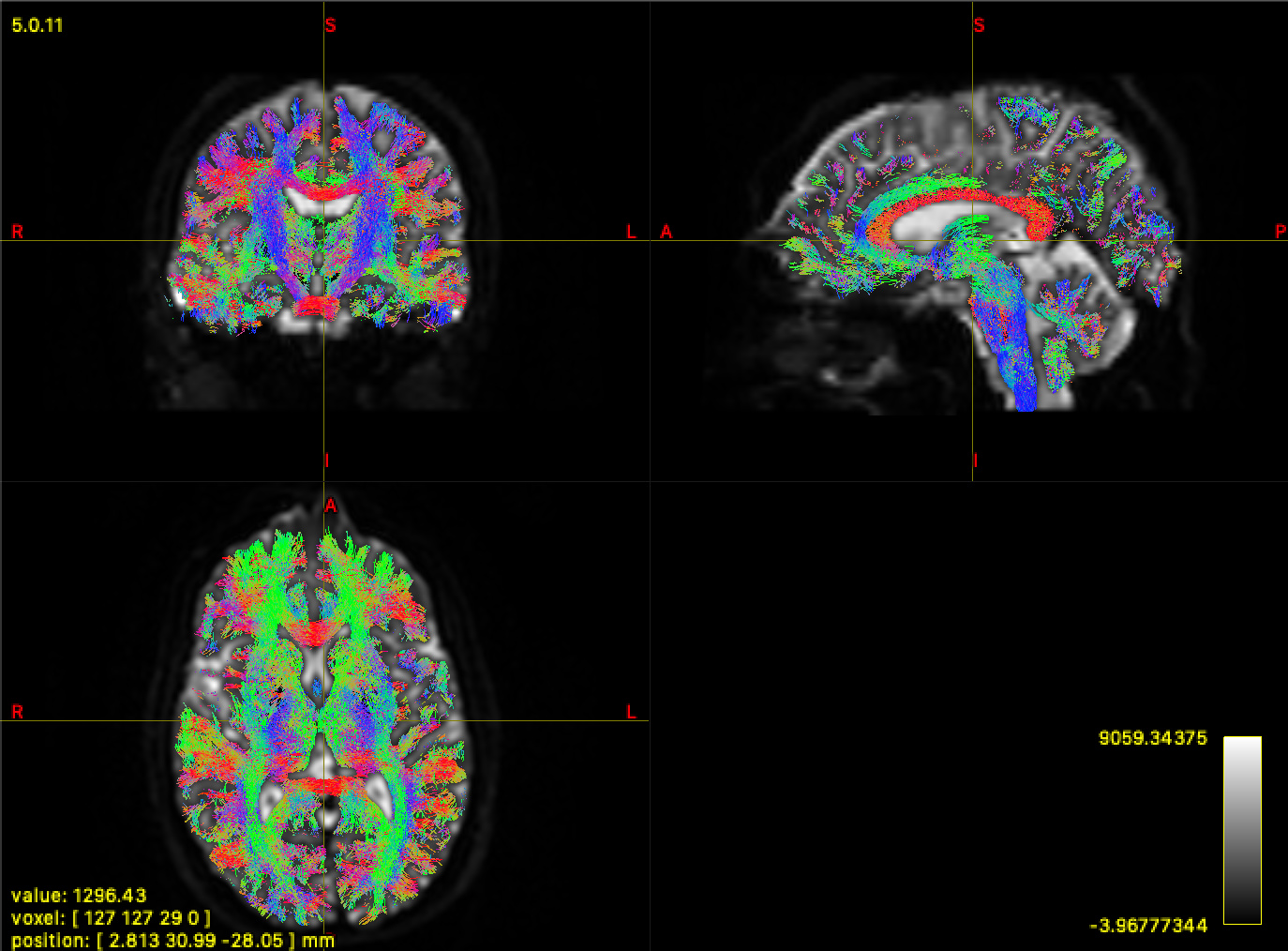
The previous iteration of my pipeline only calculated WM and GM ODFs, but I’ve changed it due to recommendations on the forum. Yet, the tractography from that iteration and the WM FOD looks much better. Here is the WM FOD:
What is going on? Should I stick with my original pipeline? The only differences were the WM/CSF vs WM/GM and adding global intensity normalisation, but I don’t believe that would make such a change… Do I need a white matter mask in my tcksift2 command? Right now I’m simply feeding the DWI mask to -proc_mask.
The preponderance of streamlines in the GM will be a combination of:
Your lower b-value, where the mean DWI intensity is greater in GM than in WM; hence your “WM” FOD magnitudes are bigger in GM than they are in WM, making terminating streamlines using FOD amplitude quite problematic;
The reduction of the default FOD amplitude cutoff threshold in version 3.0_RC3; see this discussion. The new default cutoff is perhaps more tailored for a multi-tissue decomposition than single-tissue, but a compromise had to be reached as there is no trivial way for tckgen to determine from the input FOD data alone what type of model was used.
Should I stick with my original pipeline?
It’s entirely possible that in the assessment of whether use of WM/GM or WM/CSF response functions for single-shell data is preferable, the additional variable of b-value has an effect, which shifts the assessment of which is more beneficial. I don’t have experience with breadth of data that others do, and I don’t have a sense of the effect this decision might have on the intensity normalisation, but sure, it may well be that WM/GM is preferable for your data, especially if the intent is to perform tractography.
Do I need a white matter mask in my tcksift2 command? Right now I’m simply feeding the DWI mask to -proc_mask.
If you don’t provide a mask, it will automatically compute one based on voxels where the input FODs are non-zero, which should have the same result as providing the DWI mask that you used for CSD.
In the absence of a 5TT image, there’s some flexibility in what you provide here; note also that although it’s called a “mask”, the image provided doesn’t actually have to be binary, you can have values between 0.0 and 1.0. Fundamentally the purpose of that image is to have voxels where the correspondence between FOD amplitudes and streamlines densities is “less trustworthy” to make a reduced contribution toward the model fit and hence have a reduced influence on the optimisation. So if you can devise a more clever way of deriving such an image, give it a try and just see what happens (providing -output_debug might yield further insights if used in conjunction with such).
Thank you for your detailed answer. It does seem that the GM FOD also captures the CSF. I will probably stick with WM/GM for my data. If I’m ever in charge of DWI collection I’ll definitely get more shells at higher b-values…
I know I’ve played around with advising this in very very particular scenarios on the forum in the past, but I’ve updated my views on it: in most, if not all, scenarios, don’t do this. …unless maybe if you’re just after a somewhat cleaner tractography on a healthy subject. Other steps, e.g. mtnormalise in a quantitative pipeline should absolutely not be used on a WM-GM fit, in presence of genuine CSF. It’s still not perfect, but far more reasonable, to choose WM-CSF in that scenario.
In a WM-GM model, the CSF will of course be captured entirely by the GM compartment, but
Way too aggressively so: this has implications for any degree of partial voluming with CSF or other sources of free water, in that it drastically underestimates or even entirely removes the WM FOD in such voxels.
CSF has a very different b=0 signal from both WM and GM (who also differ in this regard of course, but relatively far less so). Without going into very long explanations, this is the bit that will strongly break mtnormalise’s ability to do its intended job.
In your scenario, even though it’s challenged a priori by the nature of the data, I would (in the absence of SS3T-CSD’s public availability) recommend to stick with a WM-CSF model, but change the -cutoff value (i.e. increase it beyond its default value of 0.05). While this will still be limited in its abilities to solve your observations due to…
…this is however offset a reasonable deal due to the GM being far more isotropic. Rather than a massive FOD lobe that is greater in GM than in WM, you’ll get far more noisy peaks in all directions, which spread out the signal angularly. Since the -cutoff is applied to individual amplitudes, this will help it stop a bit easier these regions; but it requires more careful tweaking of -cutoff indeed.
Seeing your observations happen more often these days is indeed a direct consequence of…
The default new cutoff might be more tailored to a 3-tissue CSD result. For a 2-tissue WM-CSF result on a dataset with a low b-value, the new default will most of the time be a bit too low.
I hope that helps and provides at least a few insights.
So the cutoff is based on the amplitude of the ODF. If I may clarify, the ODF has an amplitude for every direction indicating the density of fibers in each direction, right? So the amplitude is just the norm of this? (side question: how is ODF different from a simple tensor model?)
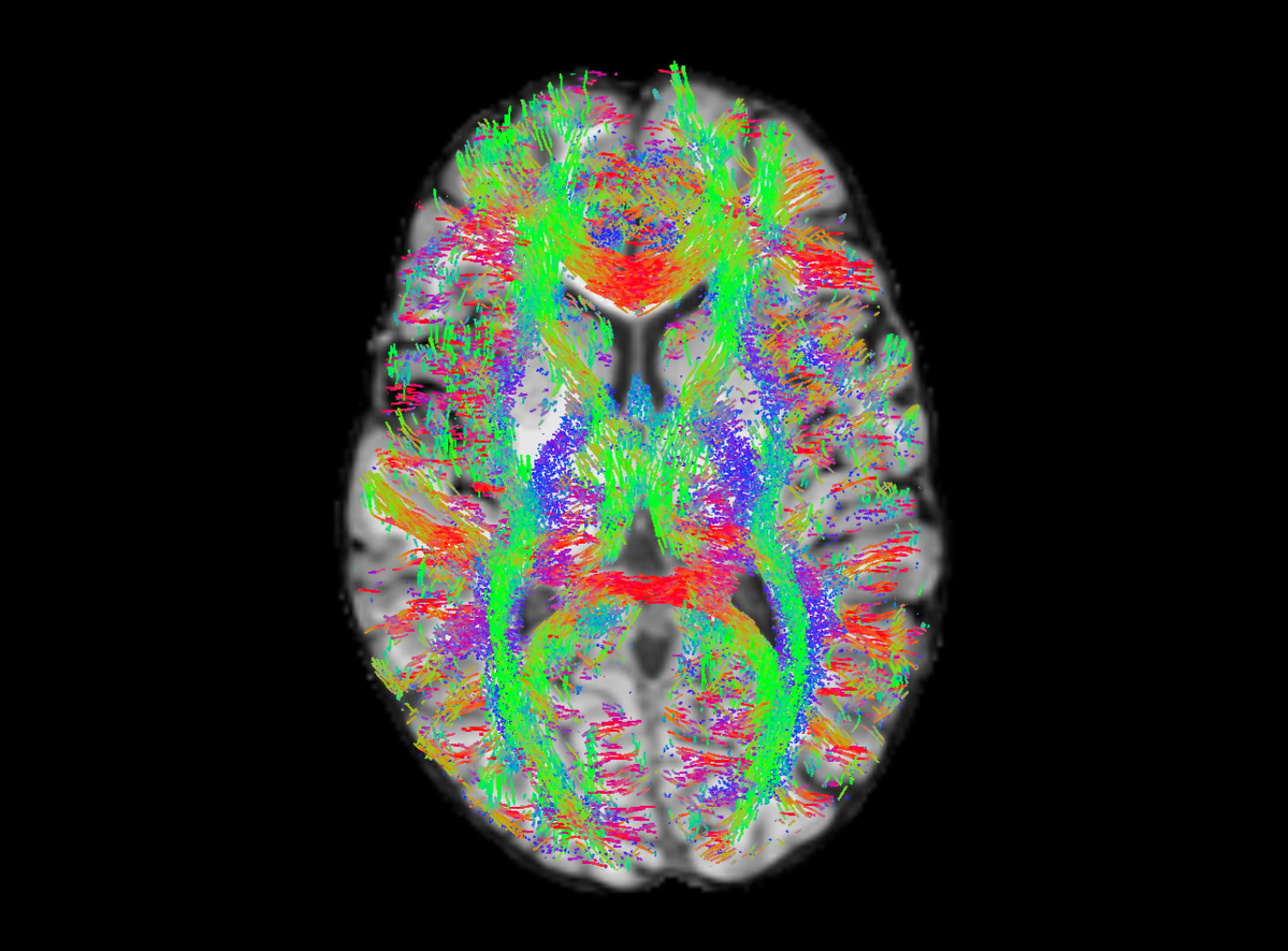
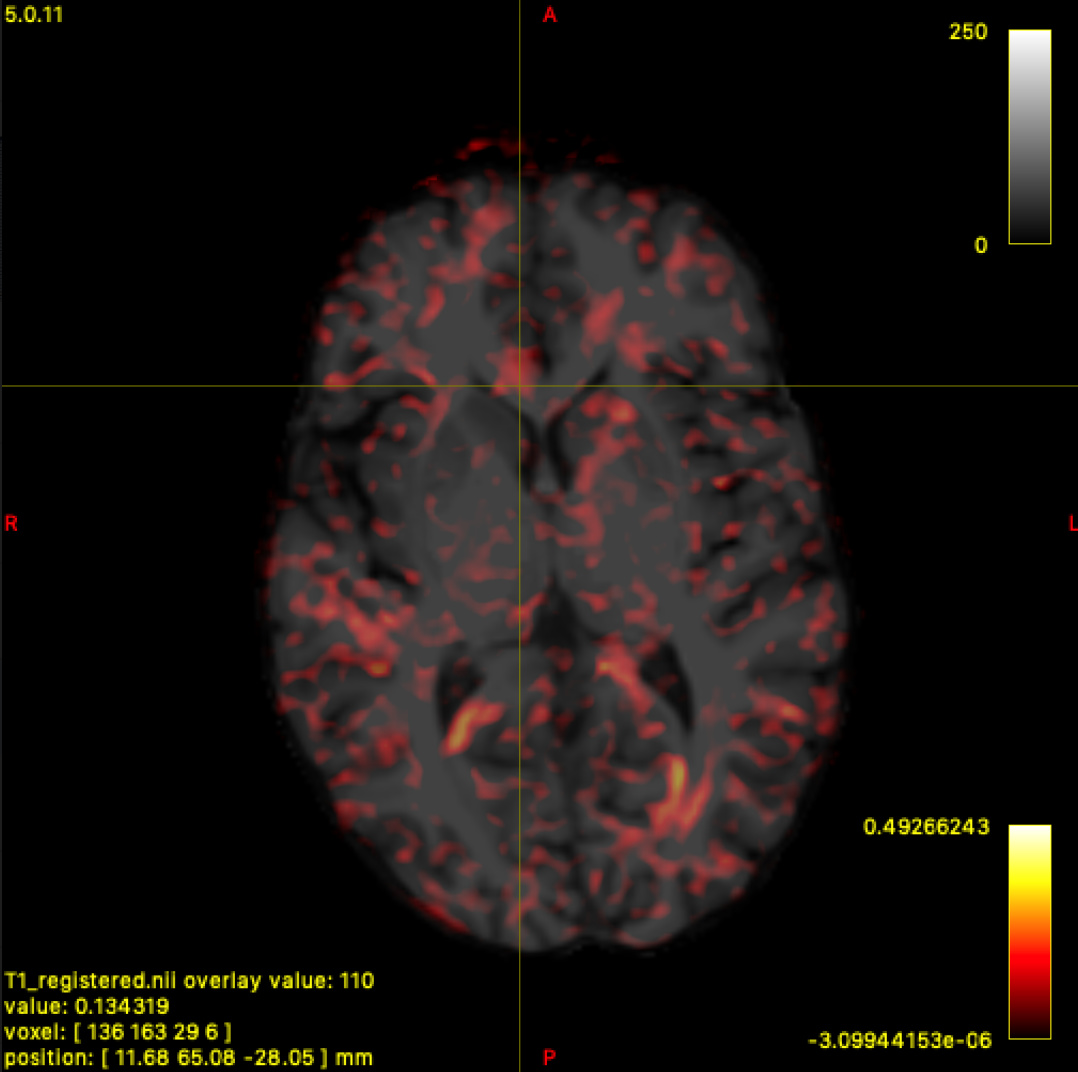
I tried varying the cutoff value and got the results below. I generated 100k streamlines at a cutoff of 0.05 (default), 0.10, and 0.15.
To me, 0.15 looks pretty solid, or maybe even in between 0.10 and 0.15. Though, I’m not entirely sure how to figure out if I’m cutting off a lot of the white matter. I tried overlay the WM FOD over the registered T1 and seeing what the values were like in the white matter for the non b0 volumes, and it does seem somewhere in between 0.10 - 0.15 seems reasonable.
Yes and no. Yep, the cutoff is based directly on the amplitude, but that is directly this amplitude for the current direction the streamline is travelling in; i.e. not the “total” amplitude of the FOD for all directions combined. It’s this total amplitude for all directions combined that is (due to your low b-value) greater in GM than WM (that’s literally that map you showed originally). The difference with the amplitude for the current direction is that the amplitude for the current direction may be lower if the amplitudes for all directions are “more spread out”, as is the case in GM. So this benefits you, as the amplitude for the current direction the streamline is travelling in will in a lot of cases effectively be lower in GM than in WM, even in your low b-value data.
That’s a whole different world. Tensor tractography is typically stopped based on FA values, which have nothing to do with amplitude (but rather with “shape” of the tensor). So no benefits or insights to be gained from trying to compare this.
Yep, generally looking at your screenshots, I more or less agree. I think 0.10 is what I would probably prefer here, even though still clearly some false positives are left in places (but it’s already far more cleaned up than the 0.05 default). So maybe 0.11, or 0.12 or something thereabouts is reasonable.
I know these ranges are “reasonable” from experience. The experience varies slightly with b-value, and type of CSD, and a few other quality aspects of the data. In practice though, it’s for each new type of data a little exploration, just like you did now, by testing a few values. To choose among those, rely on basic anatomical knowledge: if the threshold is too low, you’ll easily identify the false positives. These will reduce with higher threshold, but at some point you’ll see false negatives pop up in return. As to where to see the first false negatives “pop up” (i.e. the streamlines disappear), that would be genuine 3-way crossings, e.g. in the centrum semiovale, for the same reasons as the GM situation: more peaks means the total amplitude is divided among them, so individual amplitudes are smaller, and thus risk falling below threshold easier.
…long story short: it’s a balancing act! Once you’ve got decent choices and parameters for 1 subject though, they should translate to other subjects when using the same acquisition and processing steps.
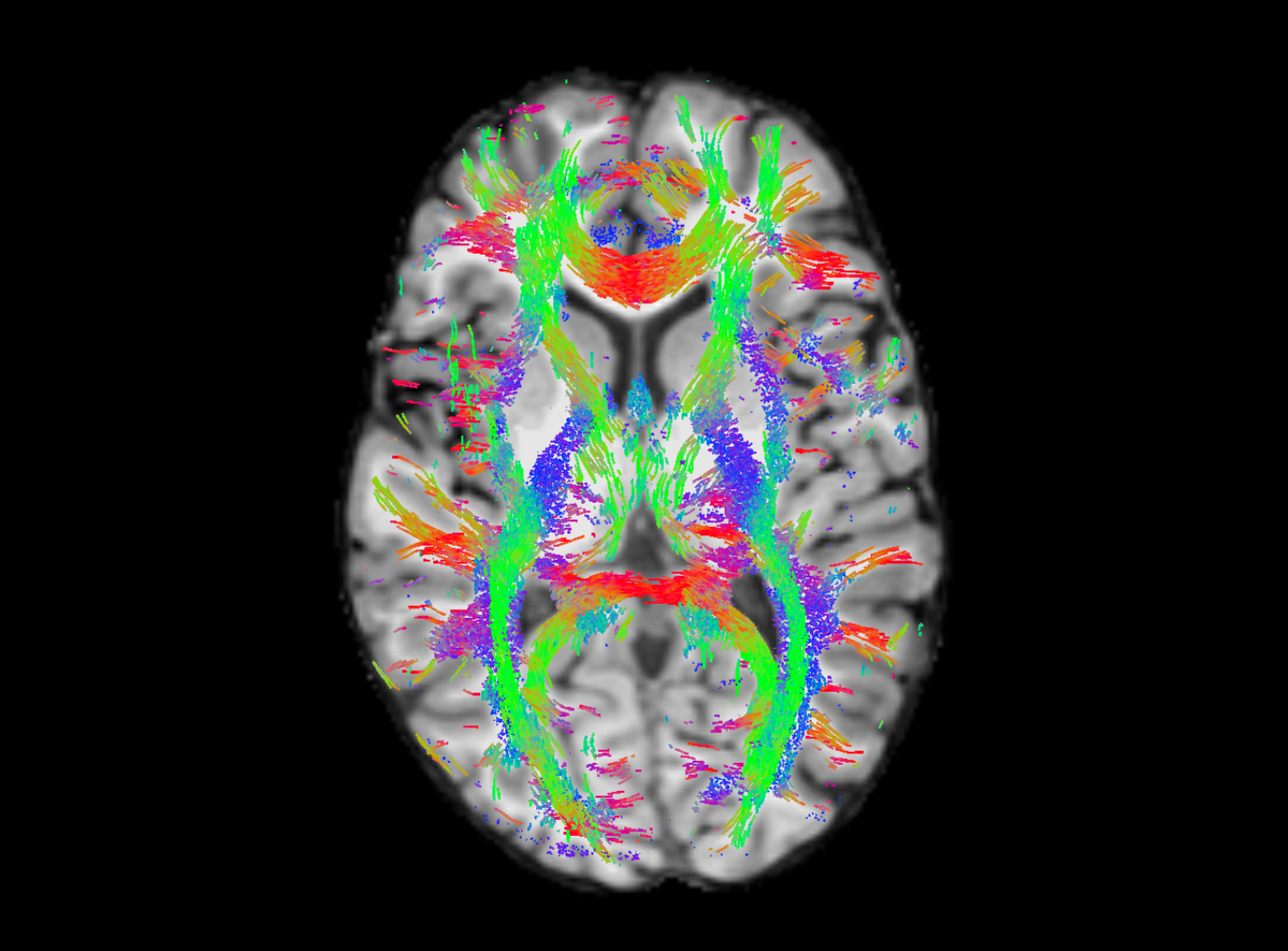
I decided to go with a cutoff of 0.12 and I got the results below. Still some false positives, but I’m thinking they will be assigned low weights in tcksift2, so it won’t affect the connectome too much. Thanks for all your help!
Based on the screenshot, that seems like a very reasonable choice.
Yep, unavoidable. Getting rid of those just by choice of threshold will almost certainly imply losing other tracts in genuine white matter. If you ever need to describe this in a manuscript or other communication in terms of “limitations of this study” kind of context, the essence is 2-fold, but only because these two points come together in this scenario:
Low b-value.
2-tissue model; essentially lack of GM compartment, but as I mentioned above, WM-CSF is still to be preferred over WM-GM in my opinion.
…and in the end, a lot of people will think this result actually still looks quite good for such a low b-value! You might want to convince your audience by effectively showing an image of the tractogram (akin to your screenshot).