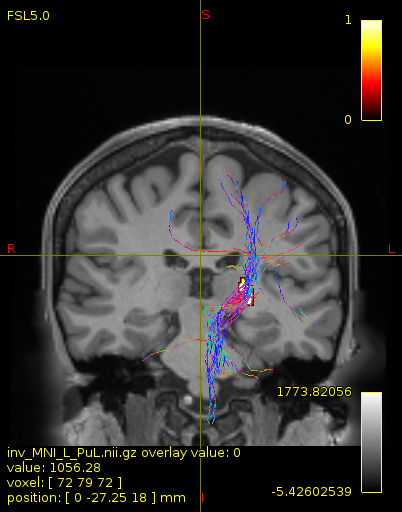
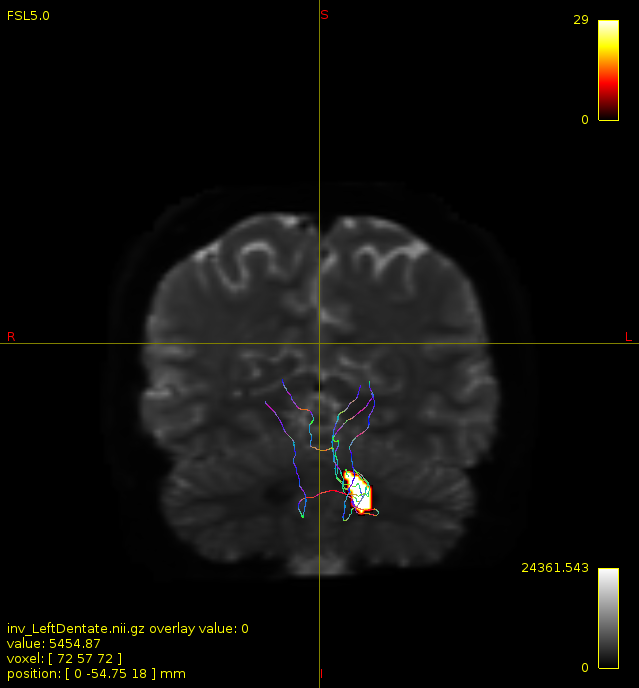
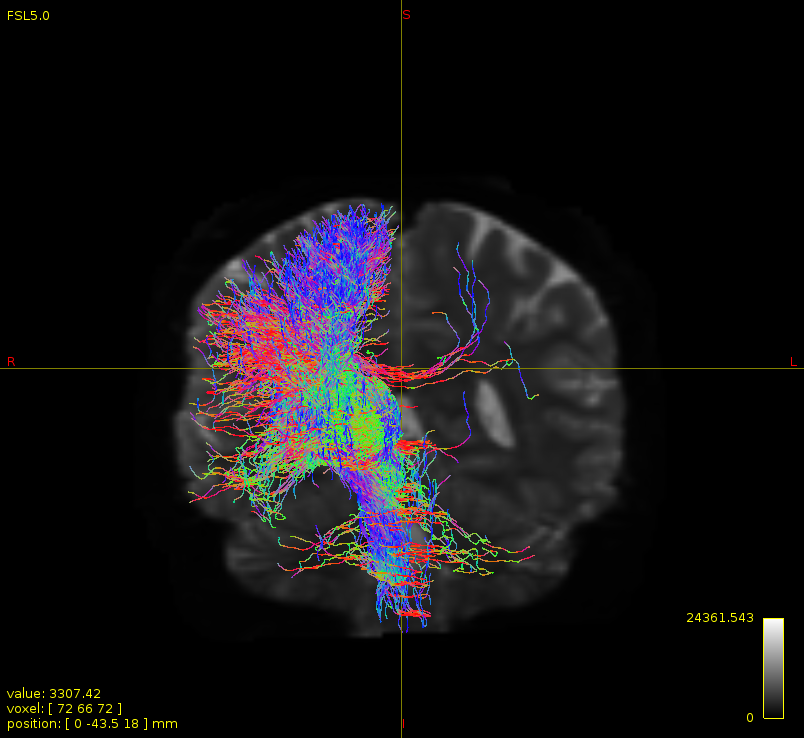
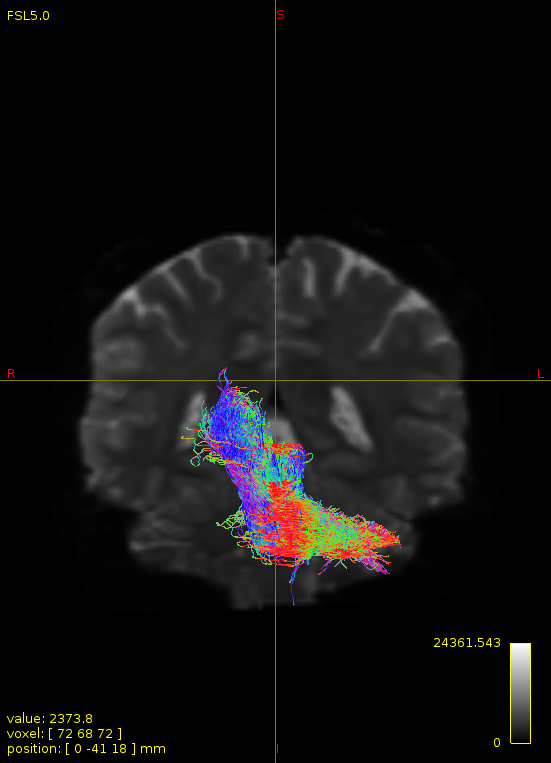
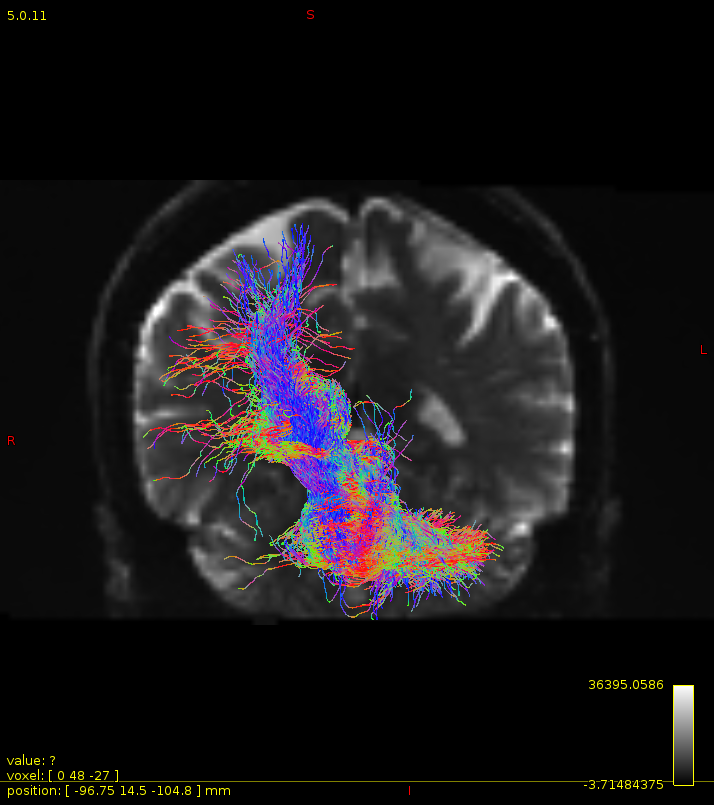
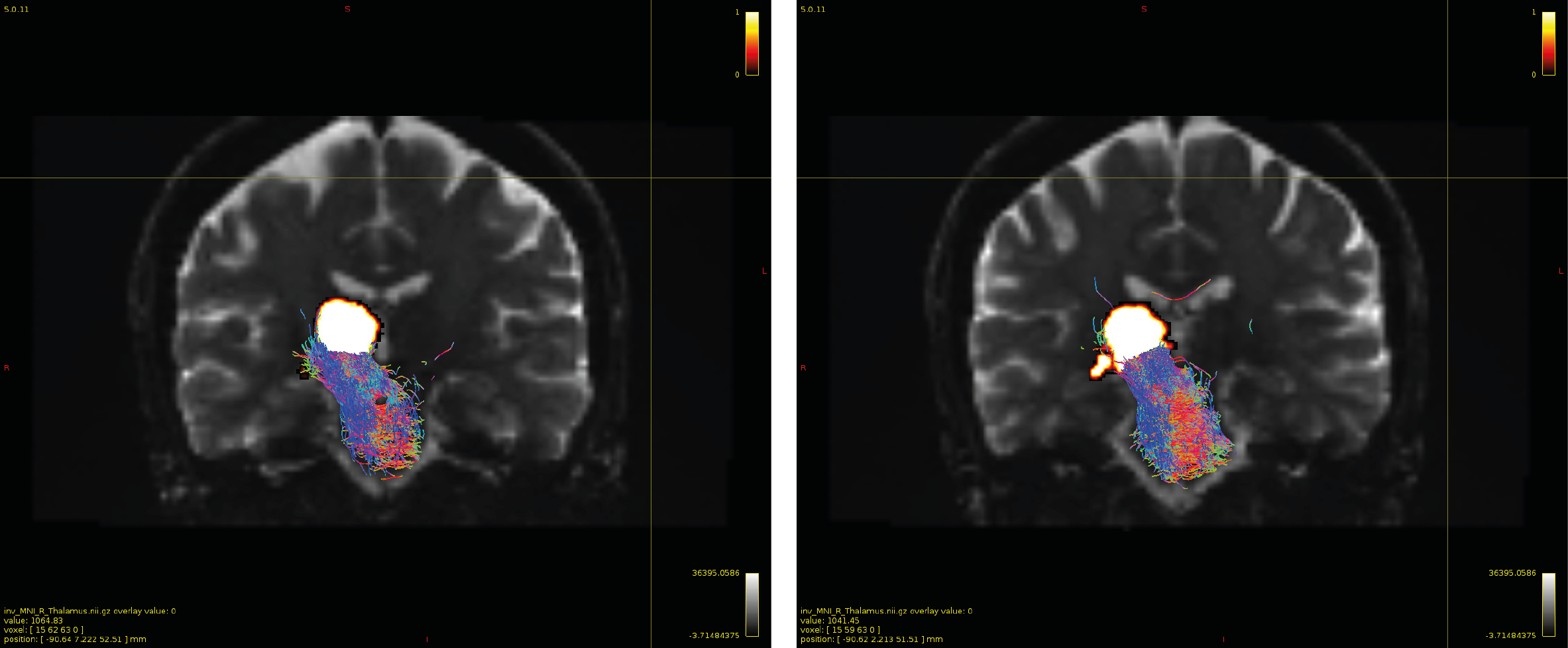
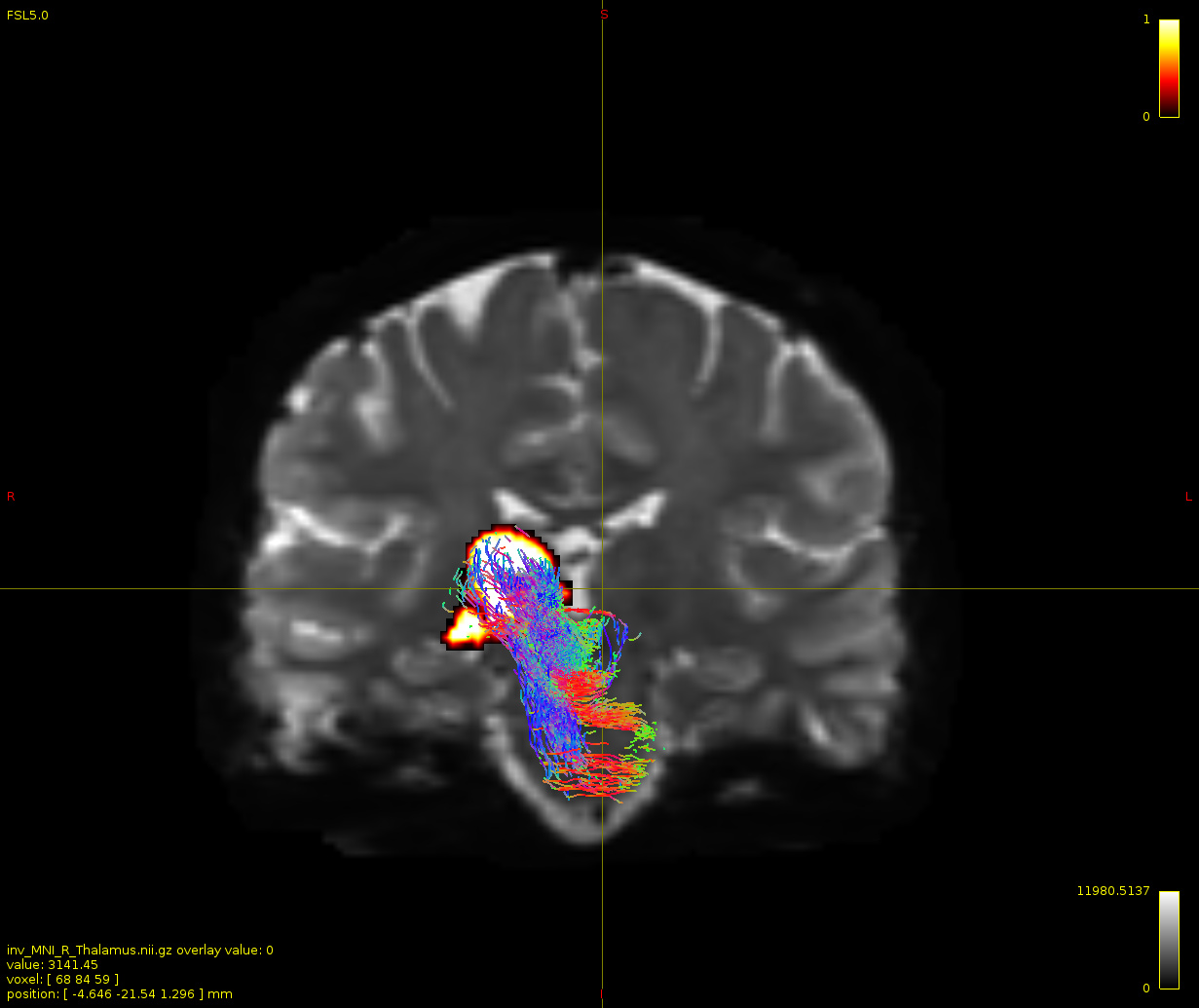
Particularly, I want to reconstruct streamlines between the dentate nucleus and the thalamus as part of cerebello-thalamo-cortical pathway and dissociate streamlines passing through different thalamic nuclei in the next step.
It’s the second half of this (emphasis mine) that I’m trying to get to more closely. I can’t provide assistance based on the names of the white matter pathways in which you are interested; but if I can understand the process that you’re trying to perform, I can provide guidance and/or alternatives.
If the using methods are not the same for both datasets, is it meaningful to compare them?
Depends on what you mean by “compare”. The fact that the datasets are so drastically different means that the degree to which you can compare the results obtained is limited, even if the underlying biology is presumably correct. So I don’t quite know how to answer this; obviously any kind of statistical comparison of outcomes can’t be done if the methods employed are not the only factor being changed between reconstructions.
Therefore, It is possible for a tract to go to the cortical regions after passing through the thalamus and not just terminate there, but the path after thalamus is not my interest.
As stated previously, this shouldn’t be happening if using ACT: once a streamline enters that region, it can’t leave. Whether or not the data exhibit this behaviour has a strong influence on how subsequent steps should be performed, so it’s important to get clarification on exactly what is going on here.
For instance, it’s possible that while it may have initially appeared as though there were streamlines being selected based on your criteria that did not terminate in the thalamus and instead projected to the cortex, this may have been in fact due to the bi-directional nature of streamlines propagation, and by limiting to unidirectional propagation, all selected streamlines would be observed terminating in the thalamus as expected.
I did not use -vector that time, I’ll try it again.
The tck2connectome -vector option is intended to be used in conjunction with tckgen -seed_unidirectional; so if you’re using the latter, the former is likely to be the preferred mechanism.
… since in creating a whole brain tractogram, we use gm/wm interface as a seed …
While GM-WM interface seeding can be used for whole-brain tractogram generation (though not exclusively; interface seeding can also be localised), it’s not the only seeding mechanism that can be used for whole-brain tractogram generation. If you’re having trouble getting adequate reconstruction density in your pathway of interest, I would suggest trying -seed_dynamic instead.
Can the inability of reconstructing the streamlines with this pipeline be explained by the low b-value?
Both the b-value and the inability to perform a 3-tissue decomposition will have an influence on the outcomes of streamlines tractography. But they don’t change the rules; they simply change the quality & attributes of the data. That’s why I’m trying to get to the heart of the underlying processing steps.
 not this time
not this time 
