I’m having trouble running dwi2response. The error message I get is:
dwi2mask: [ERROR] no diffusion encoding information found in image
myPath/mrtrix/sub10_dti64_aligned_trilin_dwi.mif;
I read here in the forum that this happens when the function is missing the gradient file, but I checked and it does exit in the same directory, as myPath/mrtrix/sub10_dti64_aligned_trilin.b. I specifically pass this when I call the function, using the -grad flag.
Any idea what might be the problem?
This is the command syntax:
dwi2response fa myPath/mrtrix/sub10_dti64_aligned_trilin_dwi.mif myPath/mrtrix/sub10_dti64_aligned_trilin_response.txt -grad myPath/mrtrix/sub10_dti64_aligned_trilin.b -lmax 4
Then it calls dwi2mask which results in the error above.
dwi2mask /myPath/mrtrix/sub10_dti64_aligned_trilin_dwi.mif /home/mayay/dwi2response-tmp-OL24KT/mask.mif
(path to the temporary directory created by the script)
I should note that I’m running mrtrix on an Ubuntu 12.04 machine, and I’m using the latest release version, 3.0_RC3 (not synced to github). When running an older release version (mrtrix3-0.3.15) I don’t have this issue and everything runs smoothly, but I wanted to update to the latest stable version which includes the bug fixes to tckgen.
I’d appreciate your assistance!
Thanks a lot in advance,
Maya
…also: if you would be unable to checkout the latest master or change your installation for some reason, you can always run an mrconvert -grad on your data beforehand, to already “pack up” the gradient table in the image file.
In general, I would even recommend this in any case (at the very earliest stage of any pipeline, i.e., when essentially importing your data): it makes all subsequent processing steps easier/shorter to specify and reduces the risk of introducing inconsistencies or other mistakes along complicated pipelines.
Thanks so much for the quick reply!
I pulled the current master from github and that did the trick.
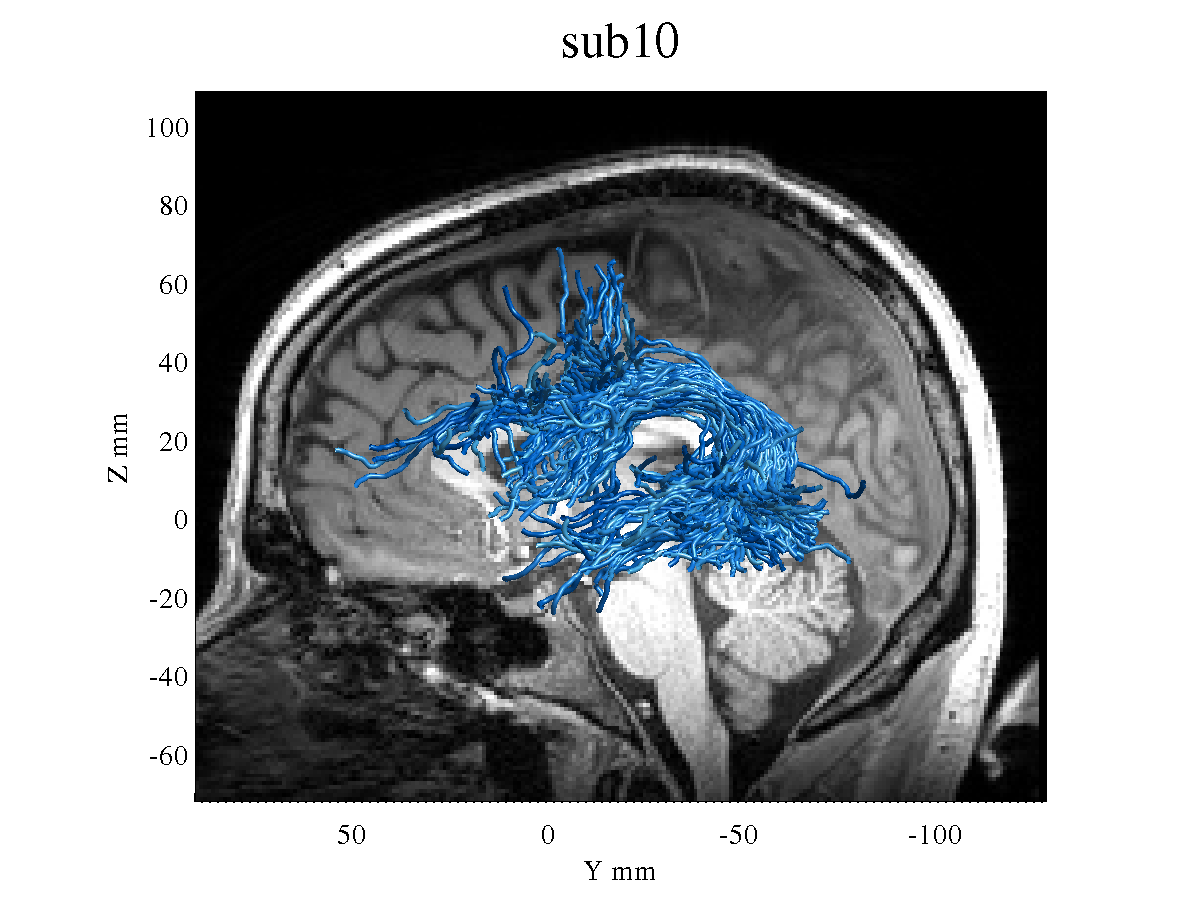
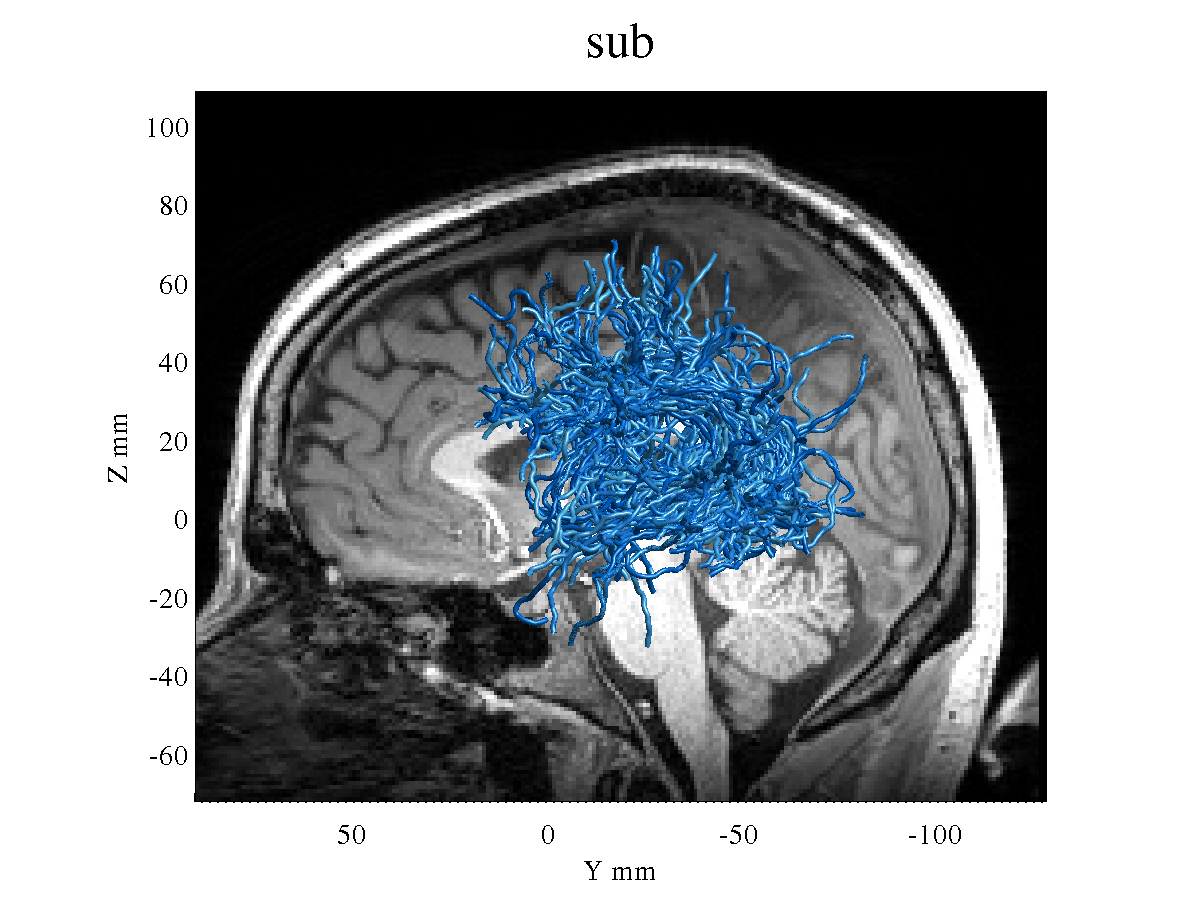
However, the resulting tracts seem very different than the ones I used to get when I was using an older release ([https://github.com/MRtrix3/mrtrix3/archive/0.3.15.zip]).
Obviously I prefer to upgrade to the latest version, but the tractography seems somehow worse, with thinner and messier tracts. See, for example, the left Arcuate Fasciculus of the same subject using the old version:
and then I used AFQ package with two ROIs to segregate the AF.
Any thoughts? Does this result seem normal to you? I know probabilistic tracking often results in fibers that are messier with many branches, but as I had previously got a cleaner looking result this is a bit worrying.
Happy to post this with more detail in a separate thread if this is appropriate.
Thanks again for the help,
Maya
This is most likely a consequence of this bug fix, and the associated changes we made at the time. It’s a long story, but in essence, the default cutoff is now more appropriate for the output of dwi2fod msmt_csd than for the regular dwi2fod csd (which I assume is what you used here?). You should be able to get close to your original results by adding the -cutoff 0.1 option to your tckgen call…
That’s right, I used the regular dwi2fod csd since I only had a single b-value in this scan. Changing the cutoff to 0.1 got me the result I was expecting! Thanks!
However, reading through the thread you referred to, I became worried that the conclusion was that a cutoff of 0.05 is more appropriate in any scenario, even though the fibers may seem less ‘pretty’ visually? Based on your simulations and experience, and given that I can’t use ACT or MSMT for this specific dataset, which cutoff would you recommend? I am not particularly interested in areas near the cortex but rather focused on the ‘core’ white matter, in the major tracts.
And this leads me to another question- in order to use csd_msmt, must all shells have the same number of gradient directions? I have scans with say 64 directions at bval=1000, 30 directions at bval=2500 and 30 directions at bval=4000- would that suffice?
Thanks again for the really helpful responses!
I would recommend you experiment with it and find what works best for your data and specific application. This is one of those things where there is no right or wrong answer. If we had really high SNR, then there would be very few noisy peaks to worry about, so we could lower that threshold. It really depends on your data and how strongly the tracts you’re trying to delineate contribute to the signal, relative to the noise. I think a cutoff of 0.1 is a good starting point - this had been the default for a good decade. But there is nothing preventing you from using a different value.
No. In fact, there’s a good argument to be made for having much higher sampling on the higher shells, since they have a lot less signal, but also a lot more useful contrast.
That would certainly be analysable by MSMT-CSD, so it would suffice in that sense. But ideally, if you have control over this, I’d recommend dropping the b=4000 shell and adding a lot more directions to the b=2500 shell - and even reducing the number of b=1000 volumes to boost the b=2500 further.
Also, bear in mind that you can use MSMT-CSD even with a ‘single-shell’ acquisition, since the b=0 volumes constitute a ‘shell’ in their own right – this means you can do a 2-tissue MSMT-CSD. Take a look at this thread for example.
Unfortunately no, we had already finished scanning, this protocol was optimized for different analyses (CHARMED)…
Thanks! I tried to follow the instructions in the linked post, and have several technical questions:
I used dwi2response dhollander to estimate the response functions. This resulted in 3 responses, even though I only have two shells (b0 and b=1000). Next, the syntax of dwi2fod msmt_csd expects 3 pairs of response+odf, one per tissue. Should I provide it with all three responses? Or only the WM and CSF? And if just the two, does the order matter?
In dwi2fod msmt_csd, should I specify l-max separately for each tissue type? I understand that the default behavior is to use the maximal lmax possible given the number of directions. So, if my data has 3 repetitions of b0 and 64 directions at b=1000, that would result in lmax=8 for the white matter?
Lastly, and perhaps not so technical: as I see it I have two alternative pipelines:
dwi2response dhollander → dwi2fod msmt_csd → tckgen cutoff 0.05
Of course I can play with the cutoff values and see what works best for my data, but just to simplify, the single shell csd will require a more restrictive cutoff, right?. In any case I will try both and look at the results, but is there any a-priori reason to prefer one over the other? In terms of accuracy of tracking, better resolution of crossing fibers, etc.?
Thanks a lot for the helpful responses!
Maya
No, just give the response and its corresponding output image one after the other, order doesn’t matter beyond that.
You shouldn’t need to. The default is lmax<=8 when the response is anisotropic, lmax=0 otherwise.
Yes, it tends to produce larger and messier fODFs, so you’d need a larger cutoff for that.
Yes, these days I would recommend the msmt_csd variant, even for single shell data. It removes a lot of spurious noisy peaks, particularly in CSF regions. Makes for cleaner tracking.