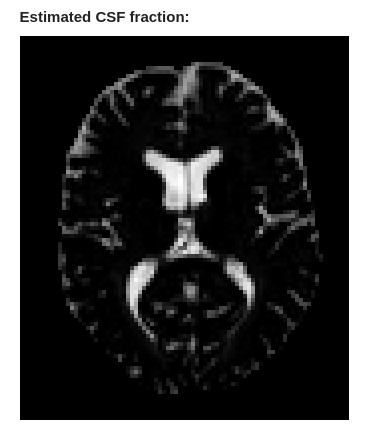
No, that’s the csf.mif image - which is the estimate of CSF “density”. As I mentioned earlier, after mtnormalise, it is close to a fraction, but scaled between 0 & 1/√4π (because it’s expressed as the l=0 SH coefficient), but not strictly so (it can go above that value, since mtnormalise doesn’t constrain the estimated densities to sum to 100% (i.e. 1/√4π), only that (after correction for the bias field) the sum of the estimated densities averages to 100%.
You could normalise the csf.mif image more explicitly as you suggest, but you will then tend to underestimate your CSF fraction since any noise in the estimated densities will produce more extreme maximum values. I wouldn’t personally recommend that.
Indeed, this is where the distinction is going to get a bit cloudy… In my mind, CSF is the closest thing to “free water” in the brain. But if you’re looking for something that estimates “free water” in the tissue, then we need to distinguish between water in the extracellular space (which can be viewed as “free” in that it is not constrained in the sense that the intracellular water is), and water that genuinely diffuses freely – which won’t generally be the case for extracellular water in healthy brain tissue, since the motion of water molecules will definitely be influenced by the cellular environment (axons, glial cells, etc).
The way the technique I’ve suggested works assumes you’re after water that can genuinely be considered to be diffusing freely, since its signal properties are assumed to match that of CSF. In general, that probably won’t capture extracellular water in the tissue. In fact, you can easily see that it doesn’t by looking at that csf.mif image itself: it will have close to zero “density” in most of the parenchyma, which doesn’t tally with estimates of 20% extracellular space that you will see in the literature.
In fact, if you really think about this, you’ll see that for healthy tissue, the extracellular water is actually already captured in the response function for each (non-CSF) tissue type. The WM response is measured from voxels assumed to be 100% occupied by WM, and their signal is used as-is as the response. The densities we obtain correspond to those that match that specific “signature” - including all of the different compartments that contribute to its signal.
As a side note: this may sound counter to the interpretation often suggested in the literature, which is that the WM odf provides an approximation to the intra-axonal water fraction, based on some of our early work. That’s because that early work only applies to single-shell CSD at high b-value – under those conditions, then yes, the odf ought to provide an estimate of the intra-axonal water (assuming healthy axons). But the analysis we’re talking about here is explicitly multi-shell, and as such, the signal at low b also contributes to the fit – and that signal will definitely contain extracellular contributions. In this context, the response functions will represent the signal you’d expect for that tissue type as a whole – including its intra and extra cellular contributions. And yes, the intracellular contribution will dominate the high b aspects of the response, but there will nonetheless be extracellular contributions in the low b range.
I guess part of the problem is I don’t know what you’re after exactly… 
Cheers,
Donald