Hi all,
I am using MRTrix to do some processing on the HCP dataset. I am testing things out on participant 100206 in the 1200 release.
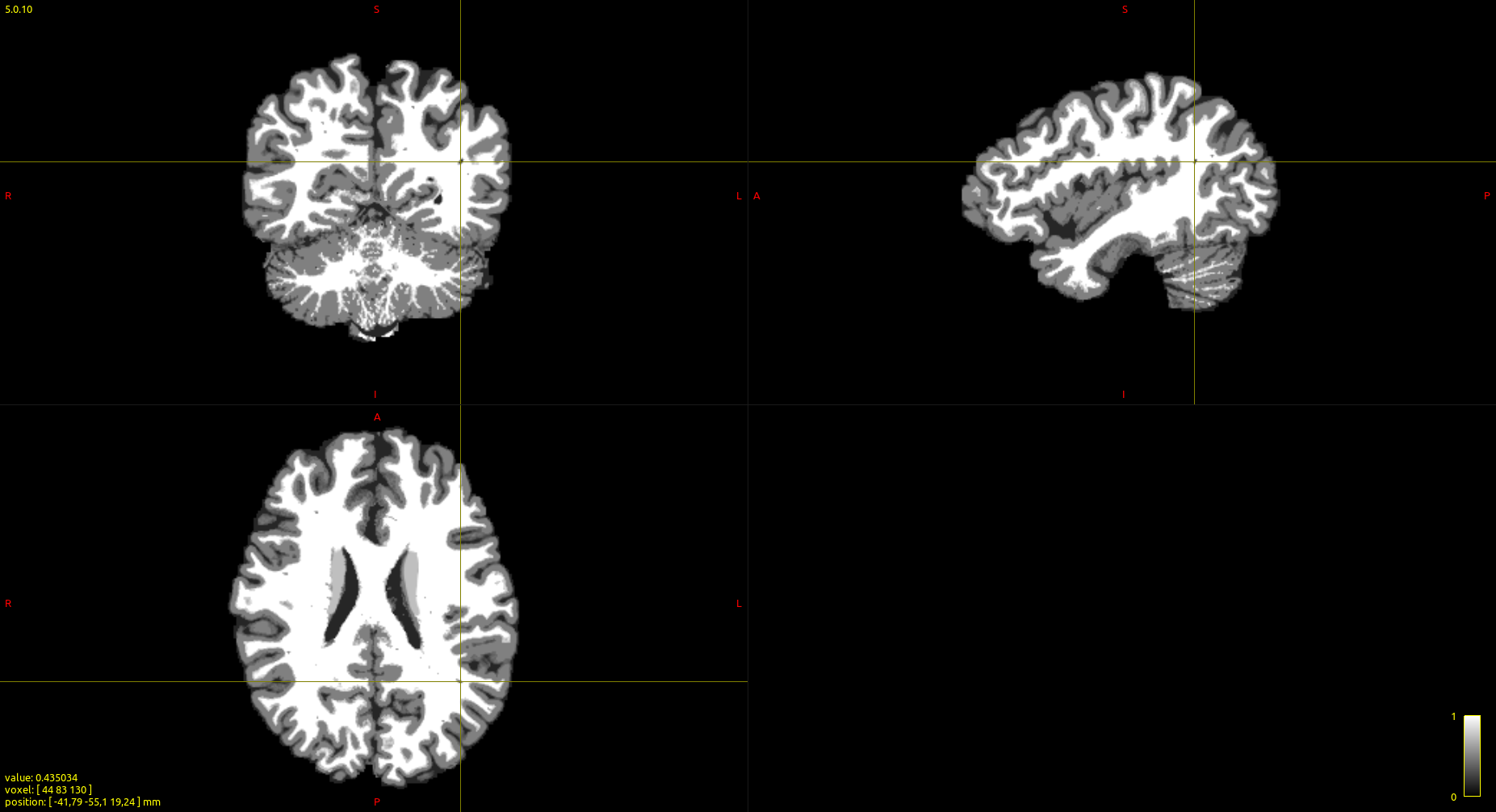
I have run the commands: 5ttgen fsl T1w_acpc_dc_restore_brain.nii.gz 5TT.mif -premasked
and 5tt2vis 5TT.mif vis.mif; mrview vis.mif
I expected vis.mif to give me specific values (as in the documentation)
Bg = 0
cgm = 0.5
sgm = 0.75
wm = 1
path = 2
However, as you can see in the attached image, some voxels are not precicely that. for example, the highlighted voxel (in white matter) has a value of 0.435.
Overall the images look rather good. Does anyone else have this experience and what do you usually do about it? Going through all participants and manually correcting all of these small errors does not seem feasible.
Because the 5TT image permits partial volume fractions for each tissue, rather than a hard assignment of each voxel to one tissue and one tissue only, the intensities in the image produced using 5tt2vis also do not necessarily have to correspond exactly to the designated intensity of one of the tissues.
For instance: Say you have a voxel that is 90% WM and 10% CGM. 5tt2vis would calculate the intensity of this voxel as:
I = (WM_fraction x WM_value) + (CGM_fraction x CGM_value)
= (0.9 * 1.0) + (0.1 * 0.5)
= 0.95
So, as one might expect, the fact that this voxel is mostly WM but with a little bit of CGM means that the resulting intensity will be almost that of WM, but pulled slightly toward the value for CGM.
If you were to generate a 5TT image that did not contain partial volume fractions (e.g. as provided using the 5ttgen freesurfer script algorithm), then in that case every voxel in the 5tt2vis output image would contain exactly one of those five values.
Also, one other little trick: When using the cursor in mrview, if you are displaying the image using interpolation (as is on by default), the value provided under the cursor will also be the interpolated value at the precise sub-voxel location of the cursor. If you wish to examine the raw voxel intensities without any interpolation, you should disable image interpolation using e.g. the “I” key; the value provided in the bottom-left corner will then behave correspondingly.
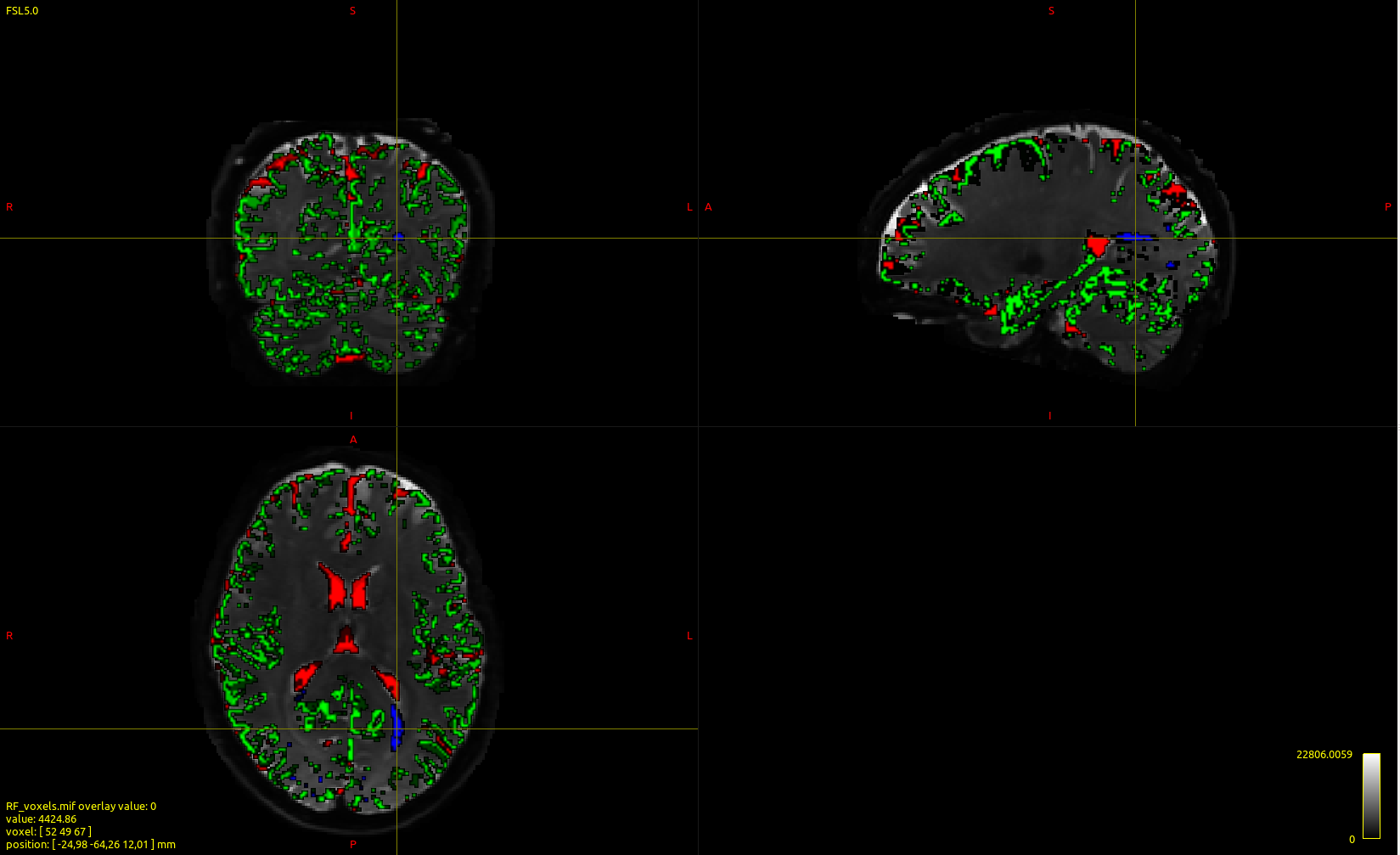
The code I used was dwi2response msmt_5tt DWI.mif 5TT.mif RF_WM.txt RF_GM.txt RF_CSF.txt -voxels RF_voxels.mif mrview meanb0.mif -overlay.load RF_voxels.mif -overlay.opacity 0.5
I was expecting the white matter voxels that are used to be in the corpus callosum proper, is it normal for the voxels used to be in the periventricular area as in the image?
I was expecting the white matter voxels that are used to be in the corpus callosum proper, is it normal for the voxels used to be in the periventricular area as in the image?
I wouldn’t say “normal”; but not unexpected. The dwi2response msmt_5tt algorithm internally calls the dwi2response tournier algorithm for selecting single-fibre voxels from a white matter mask. This algorithm selects single-fibre voxels based on the peak amplitude of the primary FOD lobe, as well as this amplitude relative to the second-largest peak in the voxel (if such exists). Due to the coil sensitivity inhomogeneity of HCP data, the posterior part of the brain is very bright in the DWIs, and since spherical deconvolution is a linear transformation (excluding the non-negativity constraint), this results in large FODs in that region of the brain, and hence selection of such voxels for response function estimation by this algorithm.
@cbajada, for future reference: the dwi2response dhollander is more accurate in general for this; there’s a mechanism in there (some sort of outlier rejection, one could say) that excludes high diffusivity values from the mask used for the tournier algorithm. Even though the single-fibre WM voxels are quite expected to come from that area, it’s still worthwhile to have some filtered out that risk coming too close to the ventricles. They may look like nice single-fibre candidates, but for multi-tissue CSD, it’s also not unimportant that they aren’t contaminated (too) much by CSF.
But for the HCP data in general, the CSF response is at risk being a bit mis-estimated by the msmt_5tt algorithm. See this quite lengthy post for more info: Dwi2response dhollander questions