Dear all
I have a dataset with half subject having a multi-shell acquisition. So I was currious to see how the new response function estimation (dhollander) perform on single shell data
I run advance preprocessing (topup bias correction) and then I run the command proposed in the documentation. First from the multishell data I just extract the first shell (64 dir at b1500)
dwi2response dhollander m4D_dwieddycor.nii.gz response_wm.txt response_gm.txt response_csf.txt -grad grad.b -mask mask_mrtrix.nii.gz -voxels sf.nii.gz -nthreads 1
dwi2fod msmt_csd m4D_dwieddycor.nii.gz response_wm.txt CSD_wm.nii.gz response_gm.txt CSD_gm.nii.gz response_csf.txt CSD_csf.nii.gz -mask mask_mrtrix.nii.gz -grad grad.b -nthreads 1
mrconvert CSD_wm.nii.gz wm_density.nii.gz -coord 3 0
mrcat wm_density.nii.gz CSD_gm.nii.gz CSD_csf.nii.gz tissueRGB.nii.gz -axis 3
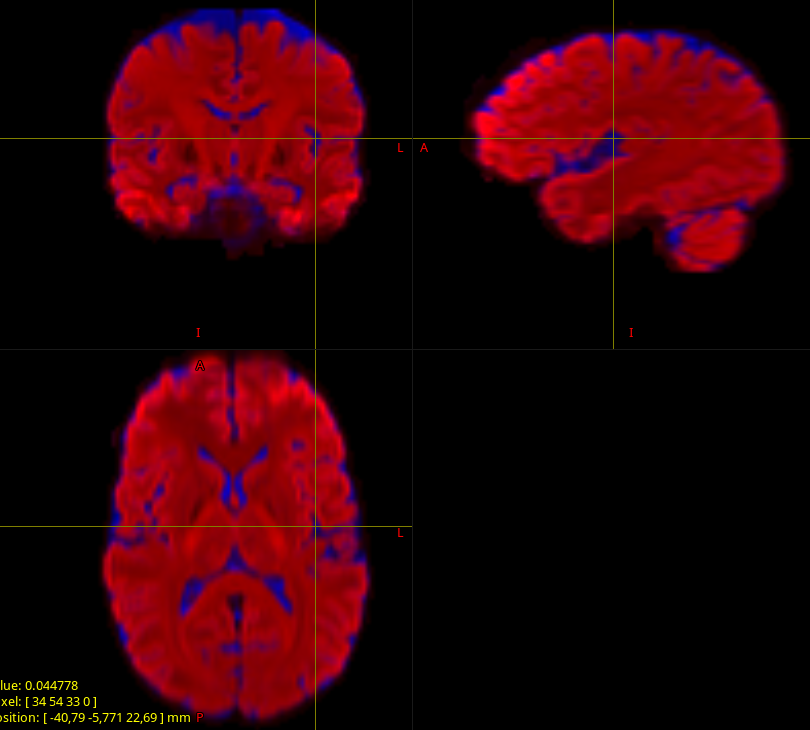
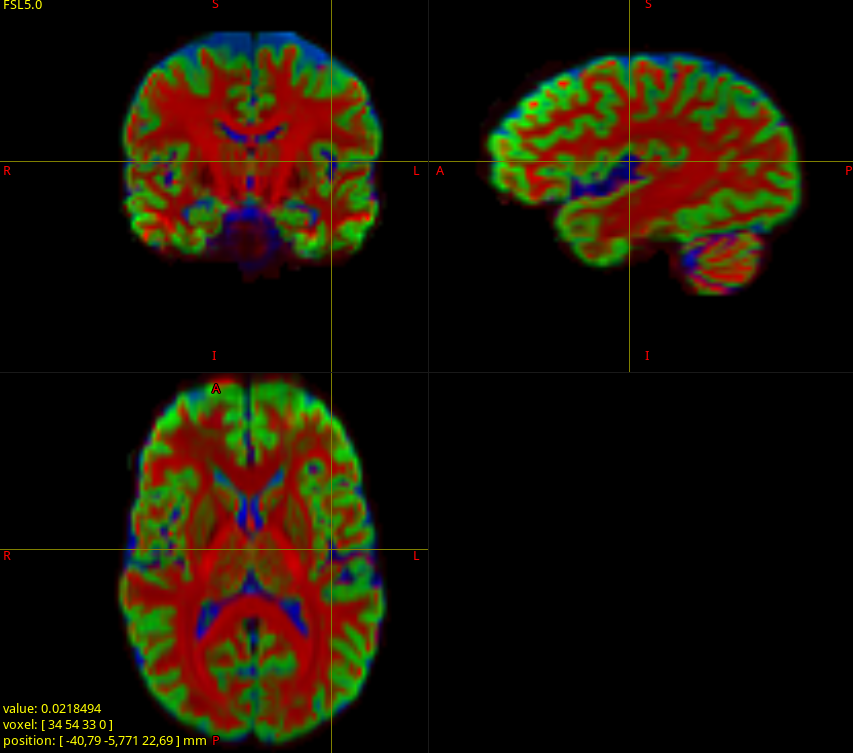
When I look to the segmented tissu map ; there is no gray matter (csf is find but white matter contains GM+WM). (This happen to all the subject)
If then I run the exact same command but this time with the complete data set (64 dir 1500, 32 dir 700 and 9 dir 300) I get a nice segmented tissue
of course this influce the white matter odf. even in the white matter.
Any idea why this happen
If usefulle here are the content of the response function :
for the single shell
cat response_wm.txt
3811.330374 0 0 0 0
1653.107621 -808.0983487 259.6307434 -52.54944092 9.225170391
cat response_gm.txt
5398.57538817
1651.10102555
cat response_csf.txt
13451.825807
244.574880543
and for the multi-shell version
cat response_wm.txt
3792.372594 0 0 0 0
2978.764202 -439.8556591 0 0 0
2341.611127 -723.8681113 121.0698332 -11.19205706 0
1652.904596 -804.2726809 257.6050592 -52.0185854 8.942642559
cat response_gm.txt
5369.25900147
4000.53468872
2864.54774623
1645.12431116
cat response_csf.txt
13496.1371533
3314.97080864
1008.28164253
330.382916373
I’m assuming when you say you extracted the first shell, you left the b=0 images in there (it certainly looks like it from the responses). Things really wouldn’t work otherwise.
In any case, the main issue here is that you’ve providing 2 shells, and trying to fit 3 tissue types. That’s not enough for multi-tissue CSD to work, it’s an under-determined problem. With the current version of the msmt_csd algorithm, you have to provide at least as many shells as tissue types.
You might be thinking of the approach that @ThijsDhollander has proposed recently, which allows processing of single-shell data and still get 3 tissue types. As far as I know, this hasn’t yet been released as part of MRtrix3 proper…
Yes I let the b=0 images
This is exactly what I am trying to test,
I used :
dwi2response with dhollander algorithm …
From what I understand this gives you the 3 response functions.
then I thought I could run dwi2fod with msmt_csd even if I get only one shell ?
or is there a part that is missing to do it right now in mrtrix ? (one should use a different dwi2fod ?)
How do I use the 3 different response function fitted with the dhollander method
can I get a tissue segmentation
Many thanks
Romain
Hi Romain,
There’s indeed a crucial confusion here (thanks for highlighting that; I’ll have to take care communicating things well in the near future!). There are two main (different) steps we’re talking about here:
- Response function estimation (
dwi2response)
- CSD (
dwi2fod)
In this work (that Donald linked to too), I proposed a new CSD method (i.e. for step 2 in the above). This method is not yet publicly available in MRtrix3 (so there’s no readily available way you can do that yet). You are correct that this would indeed be "a different dwi2fod", but when implemented properly, I’ll offer it as another choice of algorithm in dwi2fod. That will then result in dwi2fod offering 3 distinct algorithms (of which at this moment, only the first two are available):
-
csd: standard single-shell single-tissue CSD (SSST-CSD); requires single-shell data without b=0 images (the algorithm ignores b=0 images itself) and only a single WM-response; will give you a “WM FOD”, but treats all signal as if WM, so the “WM FOD” will be inappropriate when partial voluming with GM/CSF is present.
-
msmt_csd: multi-shell multi-tissue CSD (MSMT-CSD); requires data with as many distinct b-values as the number of response functions you give it (and hence, the number of tissue (FO)Ds you ask it for). The most common application/scenario is WM-GM-CSF responses, so that means you need 3 distinct b-values. Note that this includes b=0 data; some people don’t refer to this part of the data as a shell, hence some confusion around this terminology sadly. If you do apply MSMT-CSD to “single-shell + b=0 data”, and you give it 3 responses, but the data only has 2 b-values (including b=0), then you’ll get a result exactly like the one you showed. Taking into account what, GM and CSF responses typically look like, it makes sense that the GM inherently ends up not being used in the fit; i.e. the most optimal least-squares fit literally is one that has GM=0, even for hypothetical voxels that are actual 100% GM. This is the case I illustrated in the 3rd (middle) column of figure 1 in that abstract.
-
ss3t_csd (not available yet): single-shell 3-tissue CSD (SS3T-CSD); requires single-shell + b=0 data, i.e. data that have 2 distinct b-values; requires 3 response functions that have certain particular qualities relative to each other (WM, GM and CSF responses obtained from a developed human brain have these qualities, so are ok); will give you 3 tissue (FO)Ds that go with these responses. Due to the different way this optimiser is initialised and the path it traverses when looking for a decent solution, the outcome is not the same as MSMT-CSD for this case of data and responses. It will not end up with GM=0 for all voxels, but give a result more akin to MSMT-CSD on “true multi-shell” data. There’s some other particularities as well, but those go beyond most normal expected scenarios (and uncover some crucial aspects, even for MSMT-CSD).
All of these CSD algorithms need (a) response function(s) as part of the input. The algorithms in dwi2response offer several ways of obtaining them. In this work I proposed such an algorithm (i.e. the “dhollander” algorithm) that specifically looks for good WM, GM and CSF responses. It works on both multi-shell data as well as single-shell (+b=0) data, and can thus provide you with WM, GM and CSF responses suitable for both MSMT-CSD as well as SS3T-CSD. The further choice of MSMT-CSD or SS3T-CSD depends on the data you have, as explained above. The choice of dwi2response algorithm depends on what kind of responses you want:
-
fa, tax and tournier: all designed to give you just a single-shell WM response, i.e. these are the algorithms to go for if you’re looking to proceed with dwi2fod csd thereafter. fa is very dated and quite suboptimal, but included for historical reasons, and potentially still useful if you’re faced with certain very unusual scenarios. tax was supposed to be better, and also is in some cases, but can unexpectedly fail (spectacularly) too, even in a lot of quite “normal” cases. The tournier algorithm is vaguely inspired by the same iterative idea as tax, but is much more robust and would be what most/all of us would happily advise as the standard and safest go-to solution.
-
msmt_5tt and dhollander: both designed to give you WM, GM and CSF responses; both work on multi-shell data as well as single-shell + b=0 data. msmt_5tt needs an additional 5tt segmentation file as the input, which typically comes from a T1 weighted anatomical image that has to be registered to the DWI data up to sub-voxel precision, for every single voxel in the image. Not only is that a bit of a hassle, it’s also a challenge and ends up being a potential weakness for this algorithm, since it’s so very, very dependent on (the precision of) this. My intention with the dhollander algorithm was mainly to be more convenient, i.e., do away with the practical hassle of that T1 image, its segmentation and registration, undoing of all relative artefacts in both T1 and DWI data, etc… However, slightly unexpectedly, I figured out that the dhollander algorithm also ends up being more accurate. More recent tests at our end show that this is even true in the case of super-high quality data in an optimal scenario (preprocessed HCP data), and even much more so in the presence of a (certain) range of pathologies.
So in your case, if you’re looking for optimal processing of that particular data set, I’d personally happily advise you:
dwi2response dhollanderdwi2fod msmt_csd
Once the SS3T-CSD is publicly released, we could in principle speak again; however, given mostly the low maximal b-value of 1500 in that dataset, I would with 99% certainty still advise MSMT-CSD in this scenario; but not without an important warning to be a bit careful with the quantitative interpretation of the outcome. That set of quite low b-values will mean the apparent fibre density interpretation is a little bit less valid…
Hi Thijs,
thank you for this fully detailed explanation : There was indeed a confusion.
But my concern was how to process ’ single shell’ data (B0 + B1500). In the study we added multi-shell data after the start of the study. So halt of my subject have only single shell data.
This is why I want to reduce the multi-shell acquisition to a single shell in order to process the all population. (and I will use the multi-shell to see what we gain acquiring more shell, or may be for a subgroup)
So If I do well understand there is no use of the “dhollander” algorithm until we have access the the ss3t_csd method ?
So what is your advise for single shell acquisition (before the ss3t_csd becomes available) ?
Scenario 1 : reponse function with “tournier” algorithm and dwi2fod with csd.
Scenario 2 : reponse function with “dhollander” algorithm and dwi2fo with csd (and WM response estimation)
Many thanks
Romain
Aha, that clarifies a lot! So for this particular scenario, I’d actually advise what you inherently ended up doing in that first example in your first post; but given that we know the GM will never show up, you can just as well just feed dwi2fod msmt_csd only WM and CSF responses (and only ask it for WM and CSF (FO)Ds). You can see this as something close to free-water removal (the CSF compartment), and you benefit of the “hard constraint” that the msmt_csd algorithm uses (whereas the csd algorithm has a “soft” constraint, i.e., an iterative regulariser to work towards non-negativity of the FODs). Note that this is not exactly “free-water removal”, as part of the GM signal gets captured by the CSF compartment as well (but that’s ok, at least you do get rid of some GM signal in your WM FODs then). Specifically given your “low” b-value, adding CSF as a compartment can yield some benefits compared to sticking with single-tissue (WM) CSD.
See also this recent post about the same topic (even with b=1500 single-shell data as well!), and the images that were posted further in that topic (or the context prior). Long story short, this would be dwi2response dhollander followed by dwi2fod msmt_csd, but the latter only using the WM and CSF responses from the former (i.e. simply ditching the GM response).