Hi MRtrix Community,
Apologies in advance for a long post! I am a beginner MRTRIX user and I have spent a bit of time familiarising myself with this program. My primary objective is to have a workflow to go from DWI preprocessing to connectivity matrices using Freesurfer cortical segmentations to define the nodes.
I’d really appreciate it I could get some feedback/corrections on the basic workflow below (which has been written based on the tutorial), as well as some recommendations for the parameters wrt to tckgen.
T1: 1x1x1 mm3
DTI: 2x2x2 mm3
I hope this might also be helpful for other beginners learning how to construct the connectomes with MRtrix, and I will be attaching screenshots - of the same subject - for the various steps as I make progress.
Thank you all so much! ![]()
A. DWI processing
-
Process diffusion data
dwipreproc -0 $i.nii $i.mif -rpe_none -fslgrad $i.bvec $i.bval
-
Extract brain mask
dwi2mask i.mif {i}_mask.mif
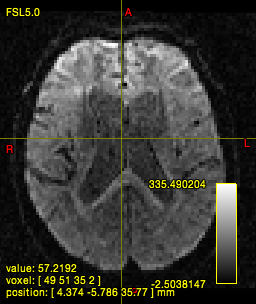
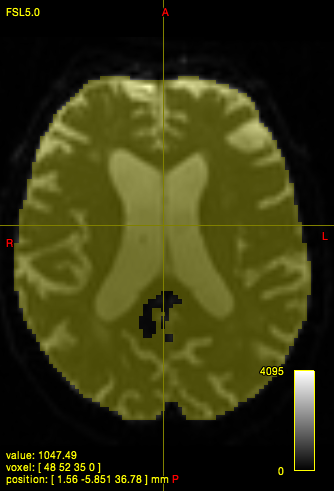
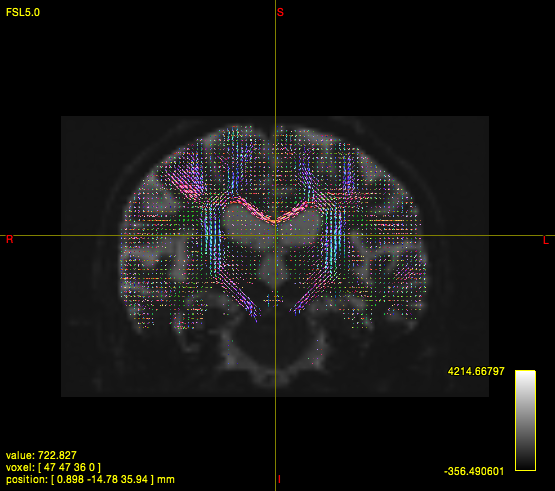
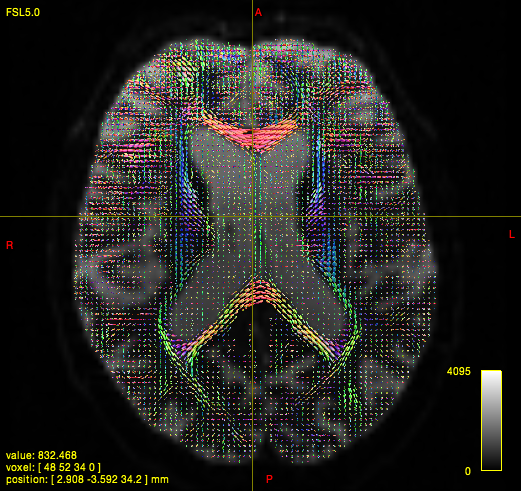
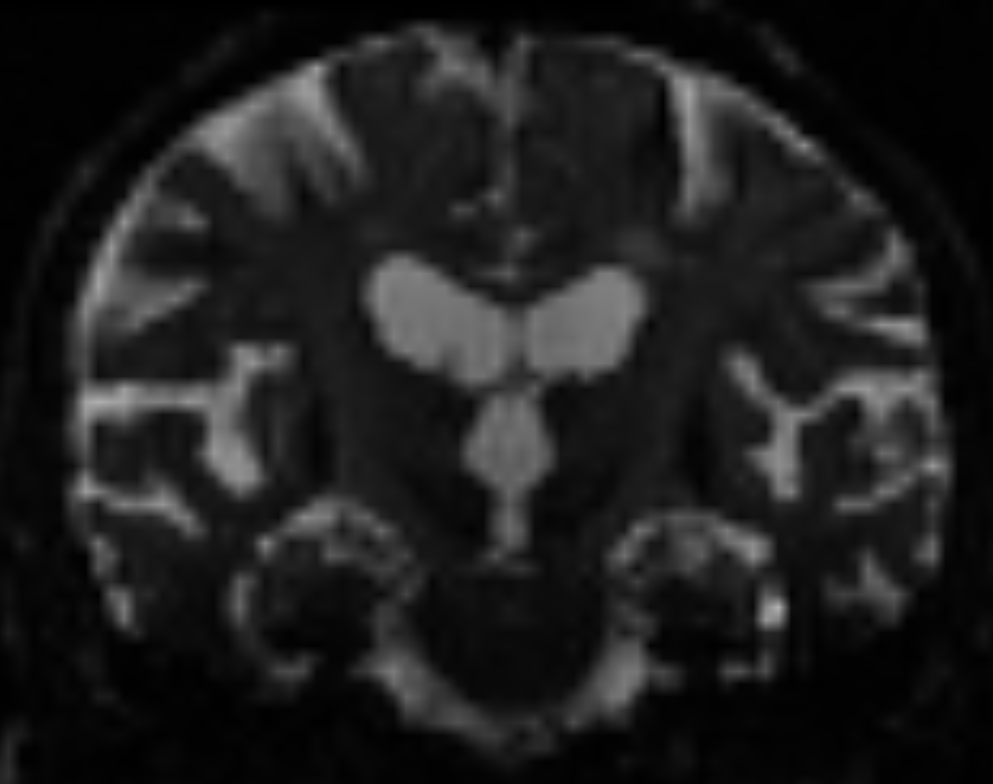
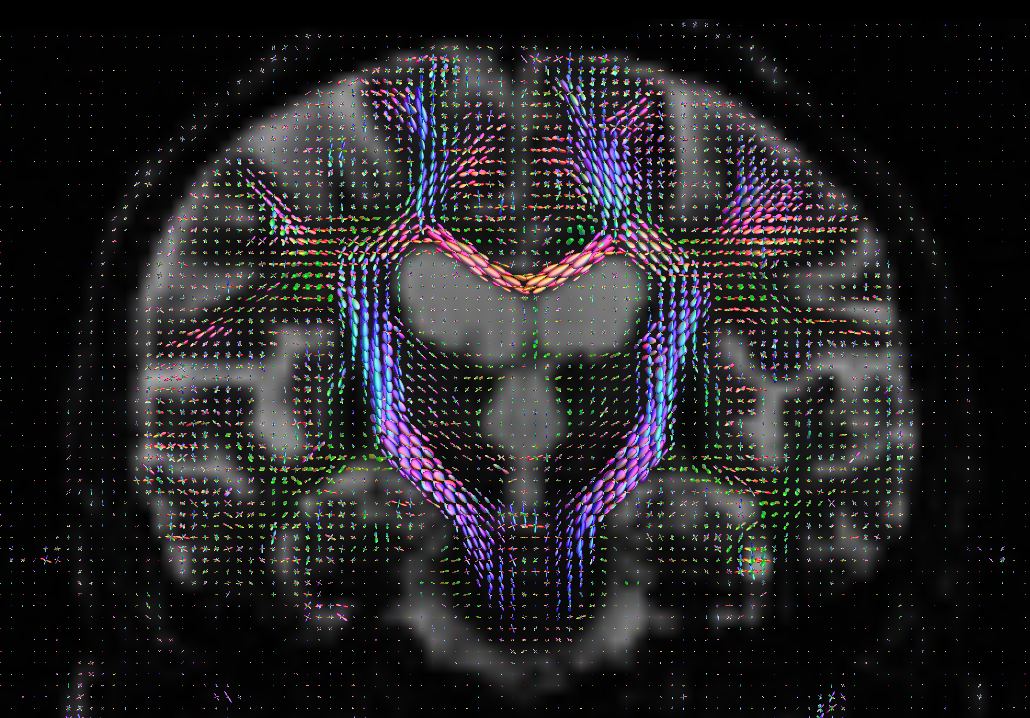
In quite a number of my subjects, I see that some of the voxels are not included, especially in the posterior regions and some subcortical structures (i.e. figures below). -
Estimated response function
dwi2response tournier i.mif {i}_response.txt
-
FOD
dwi2fod csd i.mif {i}_response.txt {i}_fod.mif -mask {i}_mask.mif

B. T1 Processing
- Process T1 in Freesurfer using standard recon-all
Result: aparc+aseg.nii.gz and T1.mgz
- Derive tissue-segmented image
5ttgen fsl T1.nii.gz 5TT.mif
QNs: Can I use Freesufer outputs here? Where does intra-individual DTI-T1 registration take place in this workflow?
- Visualise the multi-tissue image
5tt2vis 5TT.mif vis.mif; mrview vis.mif
- Modify the integer values in the parcellated image so that the numbers in the image corresponds to the rows and cols of the connectome
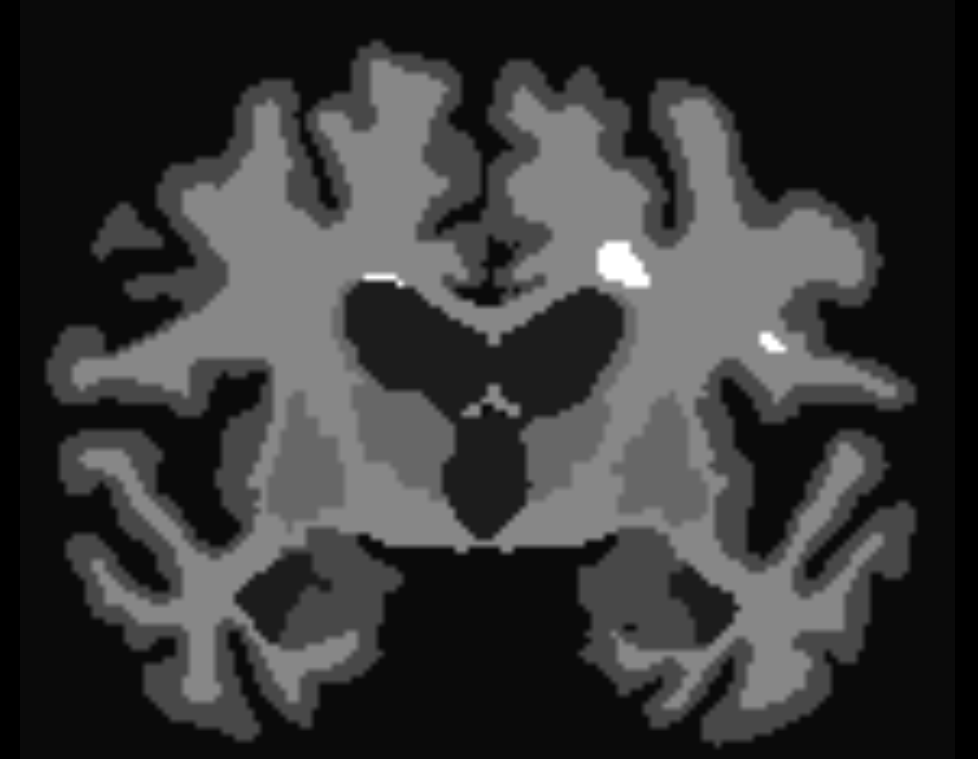
labelconvert aparc+aseg.nii.gz FreeSurferColorLUT.txt fs_default.txt nodes.mif
QN: This is assuming that I am using the Desikan atlas, right?
- Replace FreeSurfer’s estimates of sub-cortical grey matter structures with estimates from FSL’s FIRST tool
labelsgmfix nodes.mif T1.nii.gz fs_default.txt nodes_fixSGM.mif -premasked
QN: Why is this recommended in the tutorial?
C. Connectome generation
-
Streamline tractography
tckgen {i}_fod.mif {i}_tckgen.tck -seed_image {i}_mask.mif -mask {i}_mask.mif -number 1000000
-
Perform SIFT
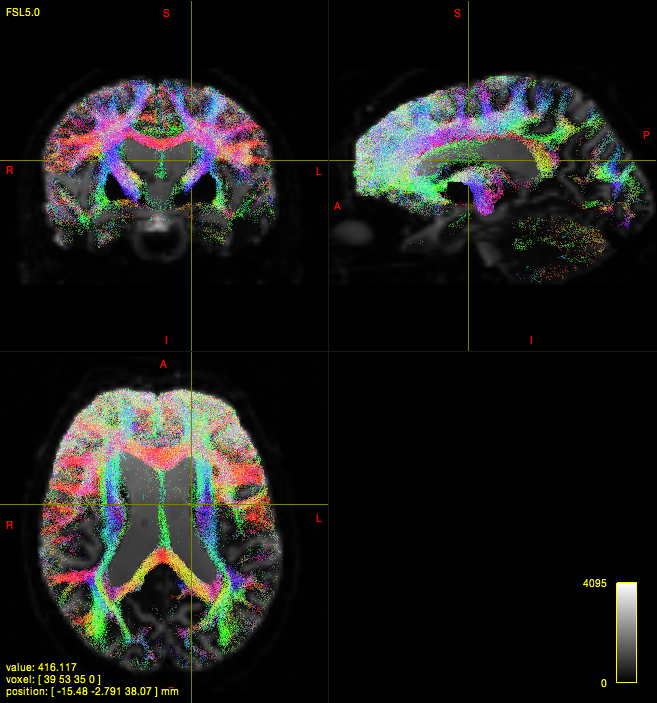
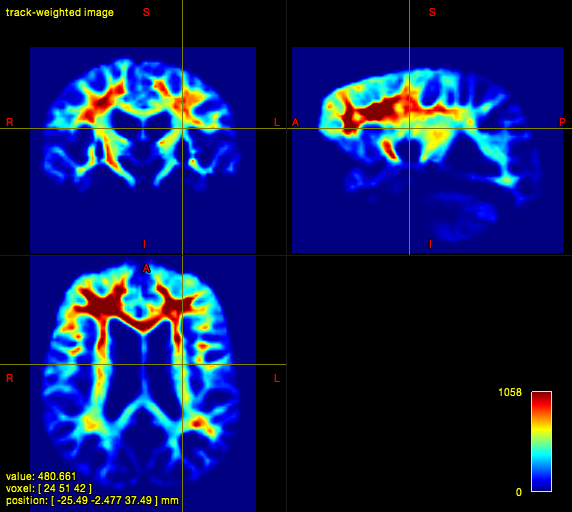
tcksift {i}_tckgen.tck {i}_fod.mif ${i}_SIFT.tck -act 5TT.mif -term_number 10M
- Map the streamlines to the parcellated T1 to produce the connectivity matrix
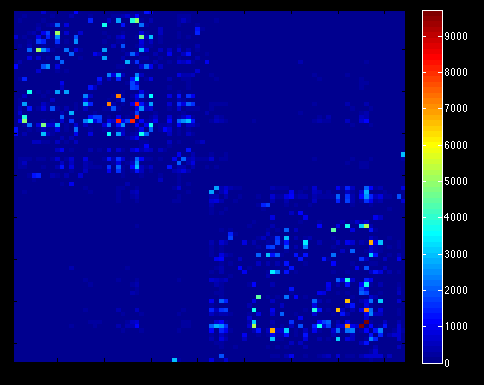
tck2connectome ${i}_SIFT.tck nodes_fixSGM.mif connectome.csv
QN: How can I extract other metrics for weighted networks such as the mean FA between 2 regions?
- View the connectome
mrview nodes_fixSGM.mif -connectome.init nodes_fixSGM.mif -connectome.load connectome.csv
- Load connectivity matrices in MATLAB for analyses with Brain Connectivity Toolbox

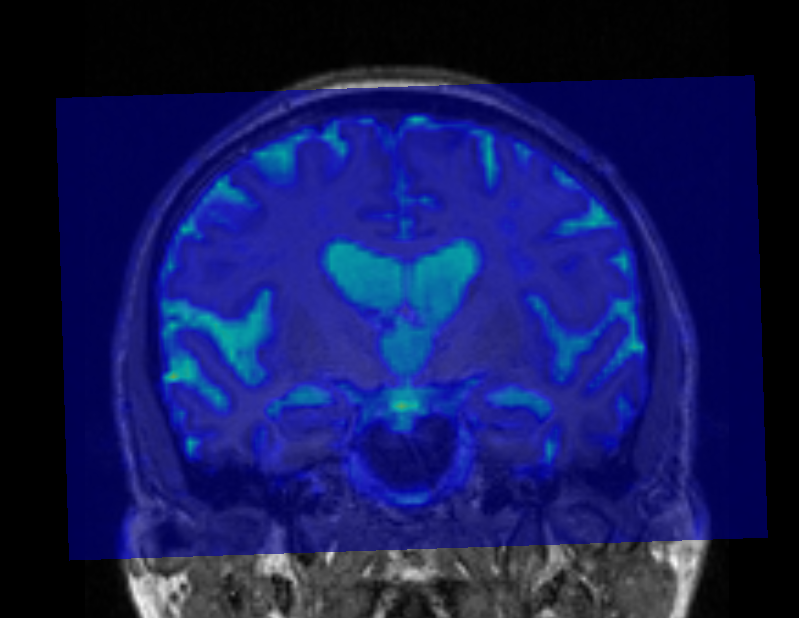


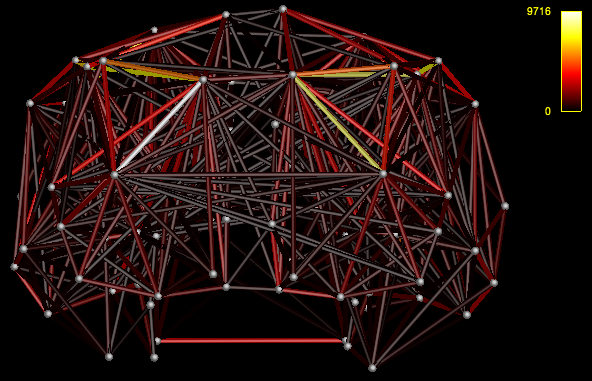
 That’s what I meant. I am sorry for the confusing wording.
That’s what I meant. I am sorry for the confusing wording.