Hi, Rob,
Thanks for your comments. I have improved the pipeline with dani’s workflow and get the connectome. I hope it would be helpful for those MRtrix beginners.
A) Segmentation(T1)
-
Cortical Segmentation (Freesurfer, 6.27 hours)
recon-all -i t1_mpr_ns_sag_iso.nii.gz -s Control_3T_zhou_yan_bert -all -openmp 8
-
Change the name of the file aparc+aseg
cp aparc+aseg.mgz ../../Control_3T_zhou_yan_aparc+aseg.mgz
-
Change the format of the segmentation file (Freesurfer)
mri_convert Control_3T_zhou_yan_aparc+aseg.mgz
Figure1:
-
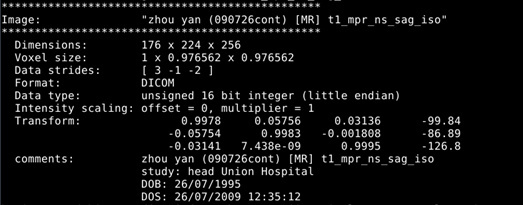
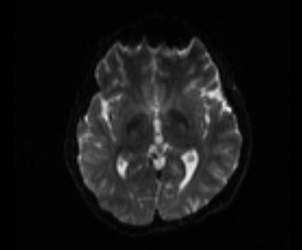
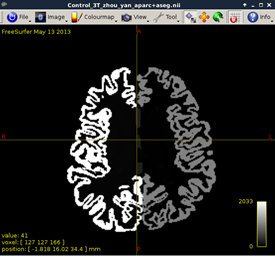
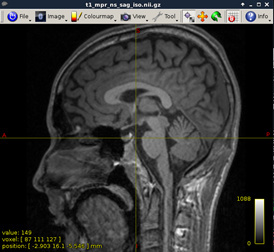
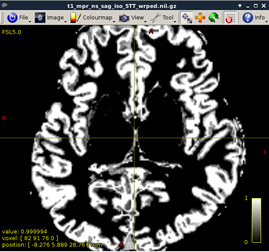
Visualize T1 data (MRtrix)
mrview t1_mpr_ns_sag_iso.nii.gz
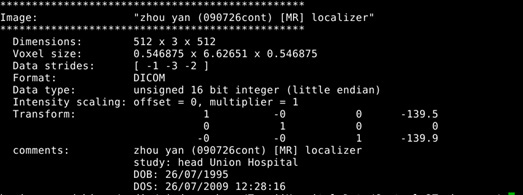
Figure2:
-
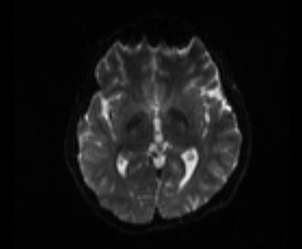
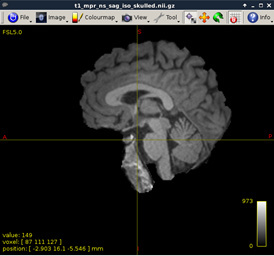
Extract the brain, because the skull can cause problem (FSL)
bet t1_mpr_ns_sag_iso.nii.gz t1_mpr_ns_sag_iso_skulled.nii.gz -R
Figure 3:
-
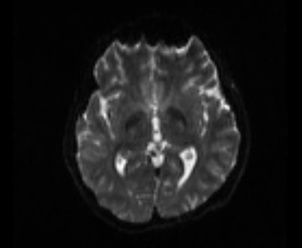
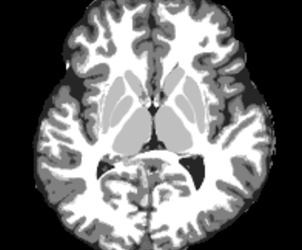
Derive tissue-segmented image (generate 5TT data) (MRtrix)
5ttgen fsl t1_mpr_ns_sag_iso.nii.gz t1_mpr_ns_sag_iso _5TT.mif
Figure 4:
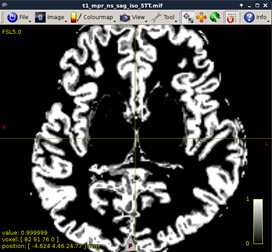
-
Visualize 5TT (MRtrix)
5tt2vis t1_mpr_ns_sag_iso_5TT.mif t1_mpr_ns_sag_iso_5TT_vis.mif
Figure 5:
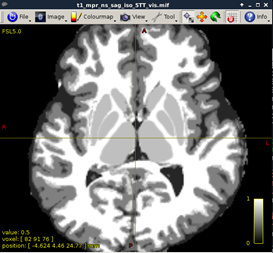
B) DWI Processing
-
Denoise DWI (MRtrix)
dwidenoise DTI_30_average-2.nii.gz DTI_30_average-2_denoise.mif
Figure 6:
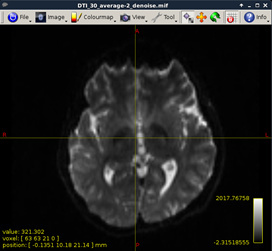
-
Preprocess DWI data (MRtrix)
dwipreproc -rpe_none AP DTI_30_average-2_denoise.mif DTI_30_average-2_denoise_preproc.mif
Figure 7:
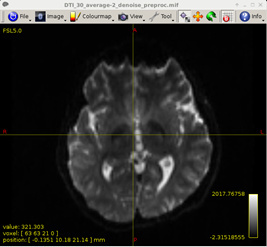
-
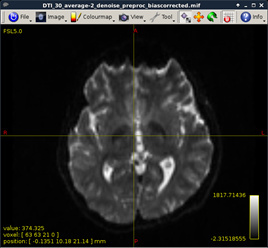
Inhomogeneity correction for DWI data (MRtrix)
dwibiascorrect DTI_30_average-2_denoise_preproc.mif DTI_30_average-2_denoise_preproc_biascorrected.mif –fsl
Figure 8:
-
Create Nift data and its bvecs and bvals (MRtrix)
mrconvert DTI_30_average-2_denoise_preproc_biascorrected.mif DTI_30_average-2_denoise_preproc_biascorrected.nii.gz -export_grad_fsl DTI_30_average 2_denoise_preproc_biascorrected.bvecs DTI_30_average-2_denoise_preproc_biascorrected.bvals
-
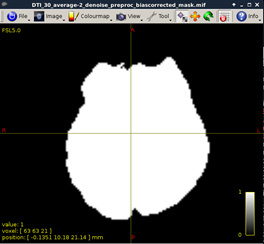
Creat mask (MRtrix)
dwi2mask DTI_30_average-2_denoise_preproc_biascorrected.nii.gz -fslgrad DTI_30_average-2_denoise_preproc_biascorrected.bvecs DTI_30_average-2_denoise_preproc_biascorrected.bvals DTI_30_average-2_denoise_preproc_biascorrected_mask.nii
Figure 9:
-
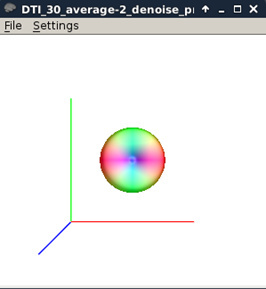
Estimate response function (MRtrix)
dwi2response tournier DTI_30_average-2_denoise_preproc_biascorrected.mif DTI_30_average-2_denoise_preproc_biascorrected_response.txt
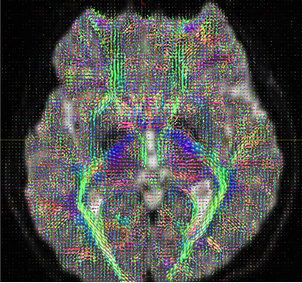
Figure 10:
-
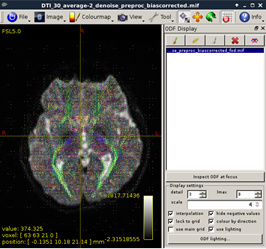
Obtain the Fiber orientation distribution (MRtrix)
dwi2fod csd DTI_30_average-2_denoise_preproc_biascorrected.mif DTI_30_average-2_denoise_preproc_biascorrected_response.txt DTI_30_average-2_denoise_preproc_biascorrected_fod.mif
Figure 11:
-
Obtain a lot of parameters(tensors), including FA, MD values (FSL)
dtifit -k DTI_30_average-2_denoise_preproc_biascorrected.nii.gz -o DTI_30_average-2_denoise_preproc_biascorrected -m DTI_30_average-2_denoise_preproc_biascorrected_mask.nii -r DTI_30_average-2_denoise_preproc_biascorrected.bvecs -b DTI_30_average-2_denoise_preproc_biascorrected.bvals
C) Registration of T1 to DWI
- align T1 to DWI (the same subject) (FSL)
`flirt -in t1_mpr_ns_sag_iso.nii.gz -ref DTI_30_average-2_denoise_preproc_biascorrected.nii -dof 6 -omat tmp_t1-dwi.mat`
-
register to MNI space (using FSL standard white matter segmentation image)(FSL)
flirt -in t1_mpr_ns_sag_iso.nii.gz -ref DTI_30_average-2_denoise_preproc_biascorrected.nii.gz -dof 6 -cost bbr -wmseg /usr/share/fsl/data/standard/MNI152_T1_1mm.nii.gz -init tmp_t1-dwi.mat -omat diff_to_template-bbr.mat -schedule /usr/share/fsl/5.0/etc/flirtsch/bbr.sch
-
transform the flirt matrix to mrtrix format (MRtrix)
transformconvert diff_to_template-bbr.mat t1_mpr_ns_sag_iso.nii.gz DTI_30_average-2_denoise_preproc_biascorrected.nii.gz flirt_import diff_to_template-bbr.mrtrix.mat
-
apply to the T1 image (MRtrix)
mrtransform t1_mpr_ns_sag_iso_5TT.mif -linear diff_to_template-bbr.mrtrix.mat t1_mpr_ns_sag_iso_5TT_wrped.nii.gz –inverse
Figure 12:
-
do parcellation (FSL)
labelconvert Control_3T_zhou_yan_aparc+aseg.nii /usr/local/freesurfer/FreeSurferColorLUT.txt /home/brain/mrtrix3/src/connectome/tables/fs_default.txt Control_3T_zhou_yan_aparc+aseg_lut.nii
D) Tractography && Connectome
-
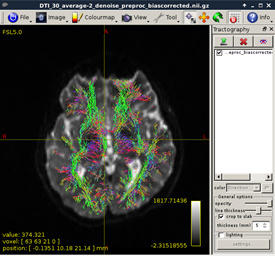
Streamline Tractography (MRtrix)
tckgen -act t1_mpr_ns_sag_iso_5TT_wrped.nii.gz DTI_30_average-2_denoise_preproc_biascorrected_fod.mif DTI_30_average-2_denoise_preproc_biascorrected_act.tck -crop_at_gmwmi -seed_dynamic DTI_30_average-2_denoise_preproc_biascorrected_fod.mif -number 20000
-
Perform SIFT (MRtrix)
tcksift DTI_30_average-2_denoise_preproc_biascorrected_act.tck DTI_30_average-2_denoise_preproc_biascorrected_fod.mif DTI_30_average-2_denoise_preproc_biascorrected_act_sift.tck -act t1_mpr_ns_sag_iso_5TT.mif -term_number 10000
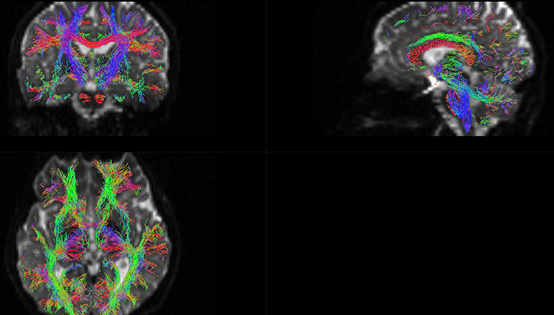
It seems very messy. I don’t know why ElijahMak can get a very nice result. Is there anything wrong when I do fiber orientation distribution? Or probably, I got a low quality dataset.
Figure 13:
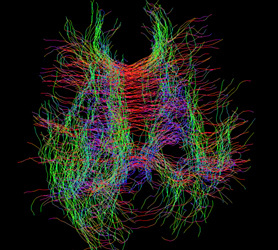
3 . tck to vtk (MRtrix)
`tckconvert DTI_30_average-2_denoise_preproc_biascorrected_act_sift.tck
DTI_30_average-2_denoise_preproc_biascorrected_act_sift.vtk`
Then I render the fibers by myself. However, it is so… ugly, so I wonder if there’s anything wrong in the workflow. The fibers seem twisted, especially the blue fibers. Also, I can not find corpus callosum fibers which should be very obvious. Do you have any ideas?
Figure 14:
4 . Connectome Matrix (MRtrix)
`tck2connectome DTI_30_average-2_denoise_preproc_biascorrected_act_sift.tck Control_3T_zhou_yan_aparc+aseg_lut.nii Control_3T_zhou_yan_connectome.csv -zero_diagonal`
I didn’t transform the matrix into a symmetric form where M is matrix
M = M + triu(M,1). For the CSV file is not compatible with Matlab with the message: “The input character is not valid in MATLAB statements or expressions”
5 . View the connectome (MRtrix)
mrview Control_3T_zhou_yan_aparc+aseg_lut.nii -connectome.init
Control_3T_zhou_yan_aparc+aseg_lut.nii -connectome.load
Control_3T_zhou_yan_connectome.csv
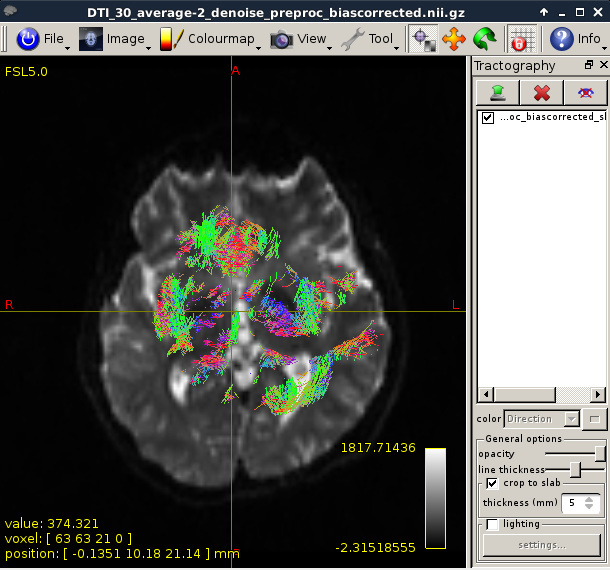
Figure 15:
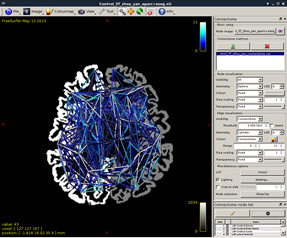
Figure 16:
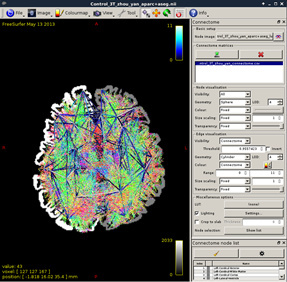
Though the connectome has been generated, I still have some questions:
① About fiber skull extraction:
As shown in current workflow, Figure 2 and Figure 3 (removed the skull) are in Left-Right orientation. Figure 2 corresponds to T1 data, and Figure 3 corresponds to T1 data(removed the skull). With T1 data, the generated fibers are shown in Figure 13, but with T1 data (removed the skull), the generated fibers are shown in Figure 17, which is obviously incorrect.
Figure 17:
② about fiber generation
Is there anything wrong with my T1 data or DWI data? I think the fibers are abnormally twisted, as shown in Figure 13 and Figure 14.
Jiangjiane has the same issue. However, I don’t quite understand what does Donald mean when he feedbacks to Jiangjiane “This is to be expected with probabilistic tractography”. Because I always thought ACT tractography is also streamline tractography. As shown in the paper, “We have introduced a novel framework for incorporating prior anatomical information into the diffusion MR streamlines tractography”
③ about registration
I think I probably have some misunderstanding of registration. I use FSL to do registration, and I follow kerstin’s steps, and your comment:“Personally I have always registered T1 to (pre-processed) DWI before anatomical image processing (including before importing to FreeSurfer);”
my current registration:
First, register the T1 data to DWI data. The transformation for this is saved as “tmp_t1-dwi.mat” in my current workflow. Second, register the T1 data to template data (here is “MNI152_T1_1mm.nii.gz”), the transformation is saved as “diff_to_template-bbr.mat”.
(register T1 to DWI, then register T1 to template, then do parcellation for T1 data)
Here is some instruction on FSL registration: “Typically, registration in FEAT is a two-stage process … …”.
My understanding:
(register DWI to T1, then register T1 to a template, then do parcellation for DWI data. Here in my data, DWI is low-resolution image, and T1 is high-resolution image)
I. Single-subject registration
Is it wrong with my registration? I am still not clear about the registration among T1 data, DWI data and Template data. What role is mrregister played in registration?
II. Multi-subject registration
If I want to do the multi-subject comparison, what should I do for registration? Does it mean I have to register all the DWI file or T1 file into the same template image?
In other words, does it mean I have to deform ( T1 or DWI image ) to (template image ), but not deform (template image) to ( T1 or DWI image )?
④ about connectome
Do the nodes of the connectome means the center of the functional area? If so, can I calculate the coordinates of the nodes to make it in the same coordinate system as the fibers?
I’m so… sorry for asking so many questions at a time. 
I hope not to trouble you a lot.
Thanks,
Sincerely,
Chaoqing