Hi MRtrix, it me again.
TLDR : see bold questions
After following through most of the tutorial for FBA (Fibre density and cross-section - Multi-tissue CSD — MRtrix 3.0 documentation) now and playing around with things a little bit, I have a few questions to make sure I’m on the right track (questions in bold so they’re easier to find in this wall of text…).
The Data
I’m using low b-value data (10 b=0 (not interleaved – they’re all at the beginning), b=1000 in 60 directions, isotropic 2.4 x 2.4 x2.4mm, all same acquisition protocol) from the same GE scanner (which unfortunately being GE, means no slice volume correction… so I was strict about removing any scans with motion). Used the most recent version of dcm2niix (GitHub - rordenlab/dcm2niix: dcm2nii DICOM to NIfTI converter: compiled versions available from NITRC) to convert all the dicoms to nifti files.
Pre-processing
I pre-processed these using PreQual (GitHub - MASILab/PreQual: An automated pipeline for integrated preprocessing and quality assurance of diffusion weighted MRI images) which did all the following: denoised (MRtrix PCA), susceptibility induced correction with SynB0 (GitHub - MASILab/Synb0-DISCO), topup/eddy correction (FSL) to fix inter-volume motion correction / eddy currents, dwigradcheck (MRtrix) to account/check for b-matrix rotations, N4 bias field correction (ANTs) to correct bias fields, and obtained a brain mask with BET2 (FSL). PreQual mentions having to set MRtrix to favour the sform NIFTI transforms over qform which they say is the default… so I fear they may be using an earlier version of MRtrix in this pipeline. So I guess potential for issues #1 if there have been important MRtrix updates since to any pre-processing commands. Potential for issues (maybe) #2: I have not yet checked whether some files are sform while others are qform… TBD.
Subject Tissue Response Functions
For 3-tissue response function estimation (dwi2response dhollander), I used the masks obtained from Pre-Qual– they fit the brain nicely + no holes (below is actually the upsampled mask but it shows the masking used).
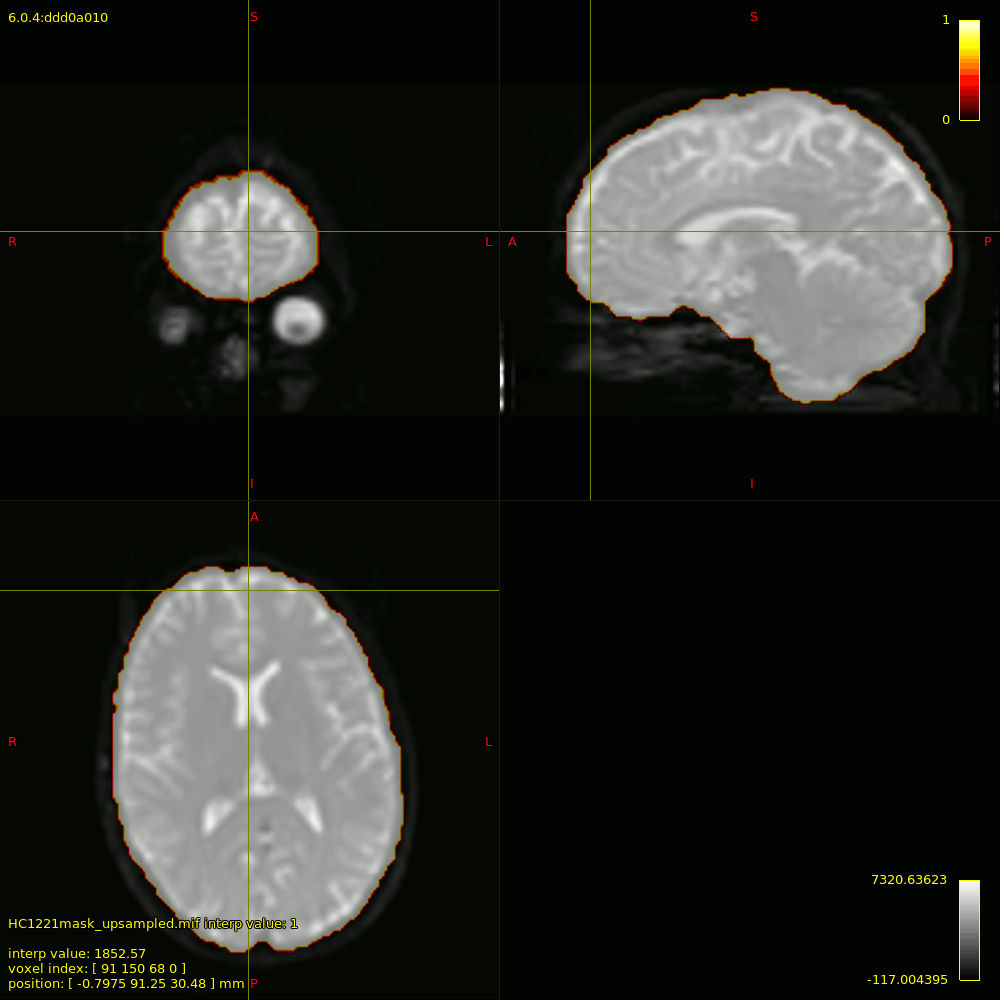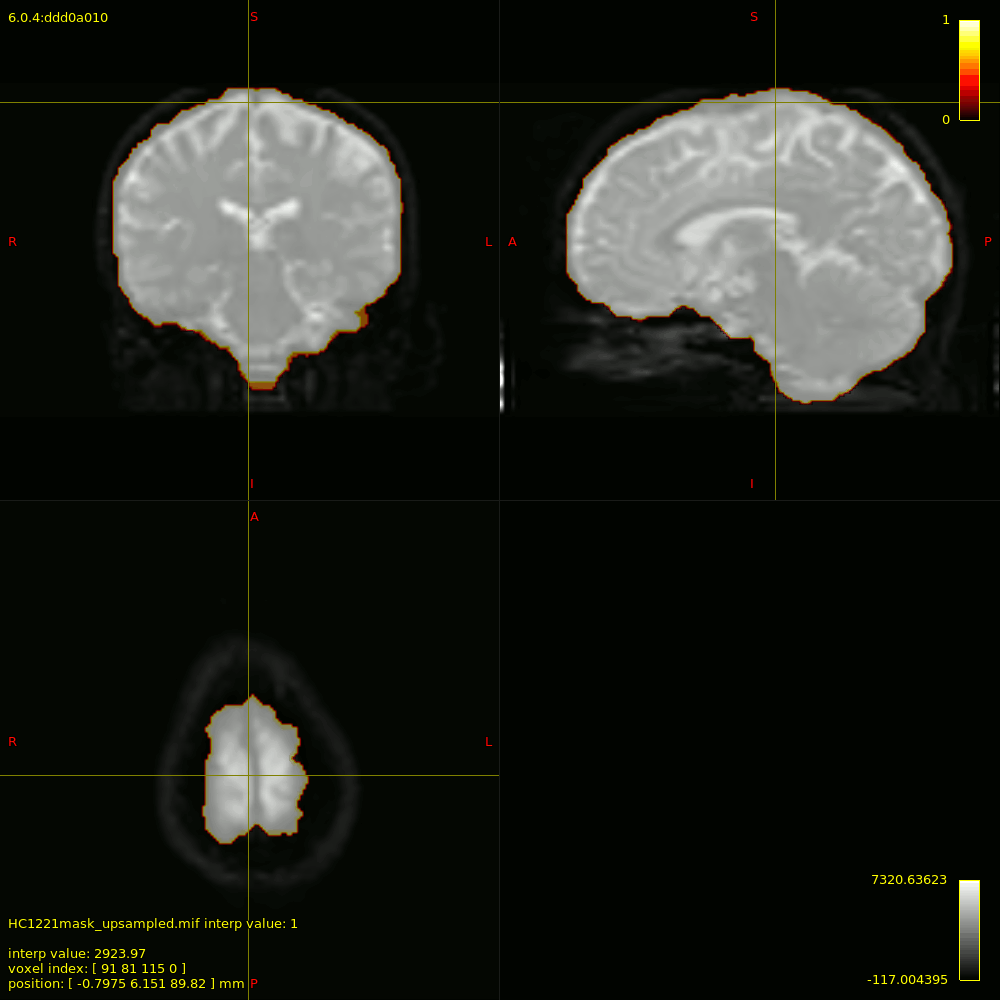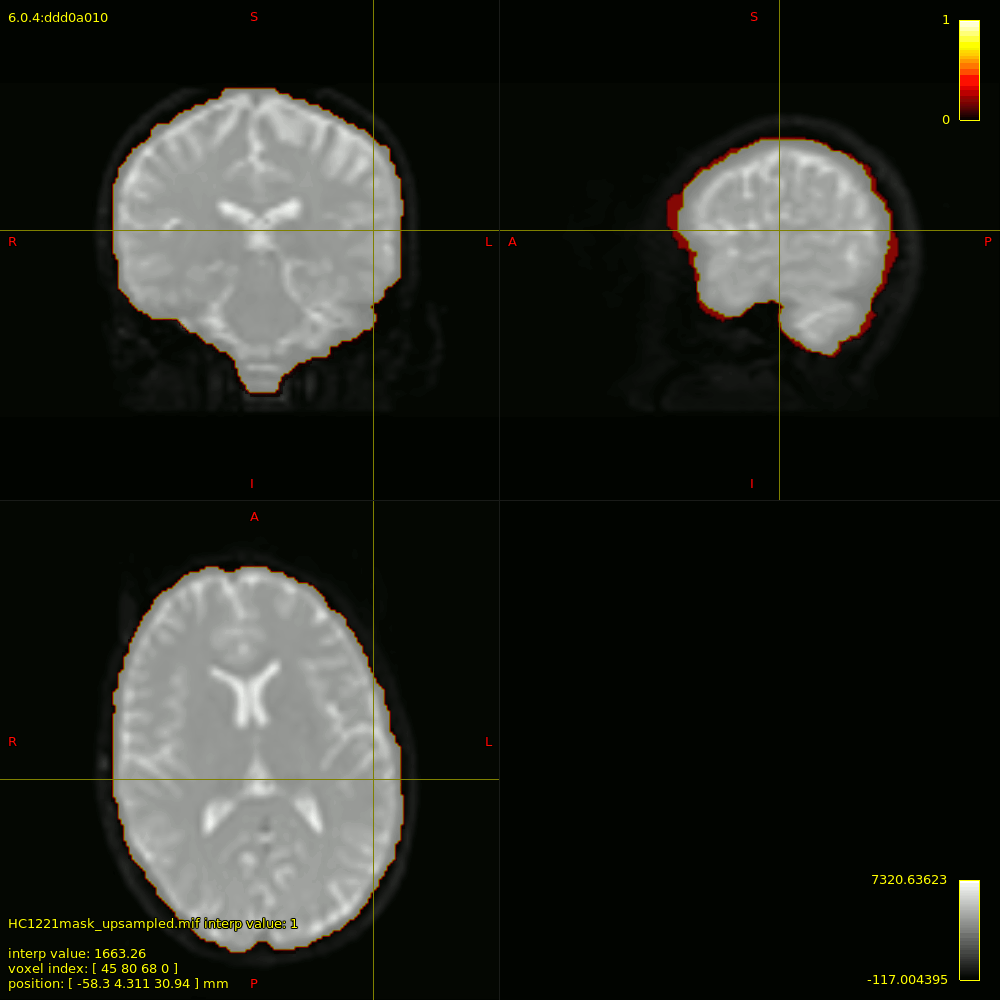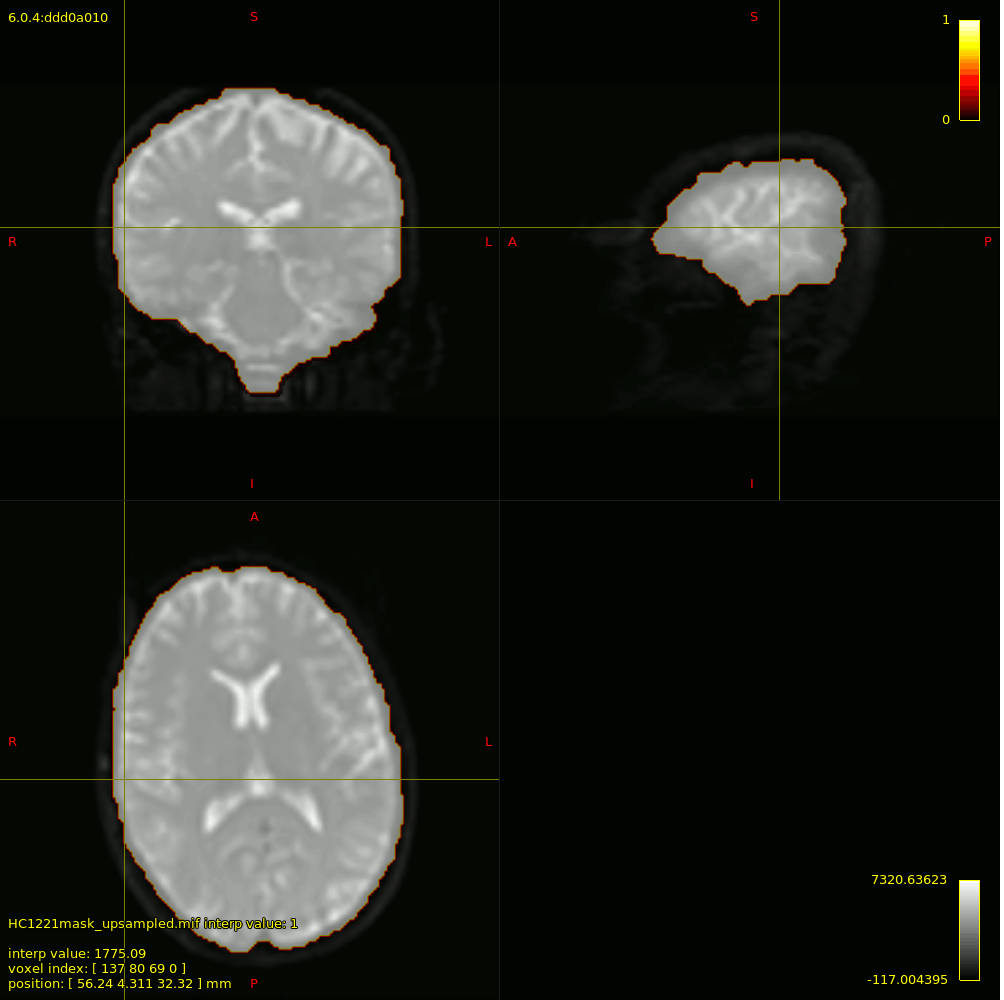
I checked the response functions with shview – they appear to be what is expected (I zoomed in a little bit on csf to show it - it was tiny).

I also checked the voxels selected for the response functions – which brings me to my first question. The sampling for GM and WM looks decent – but I notice the CSF seems to be derived largely from an area around the brain stem and not from the ventricles. Even in a participant with large ventricles, the CSF is being sampled from the brain stem area. Is this something to be concerned about? Should I be manually removing this area (I am not interesting in studying it) to prevent this area being sampled, or is this expected? The reason I ask will be clearer later on…
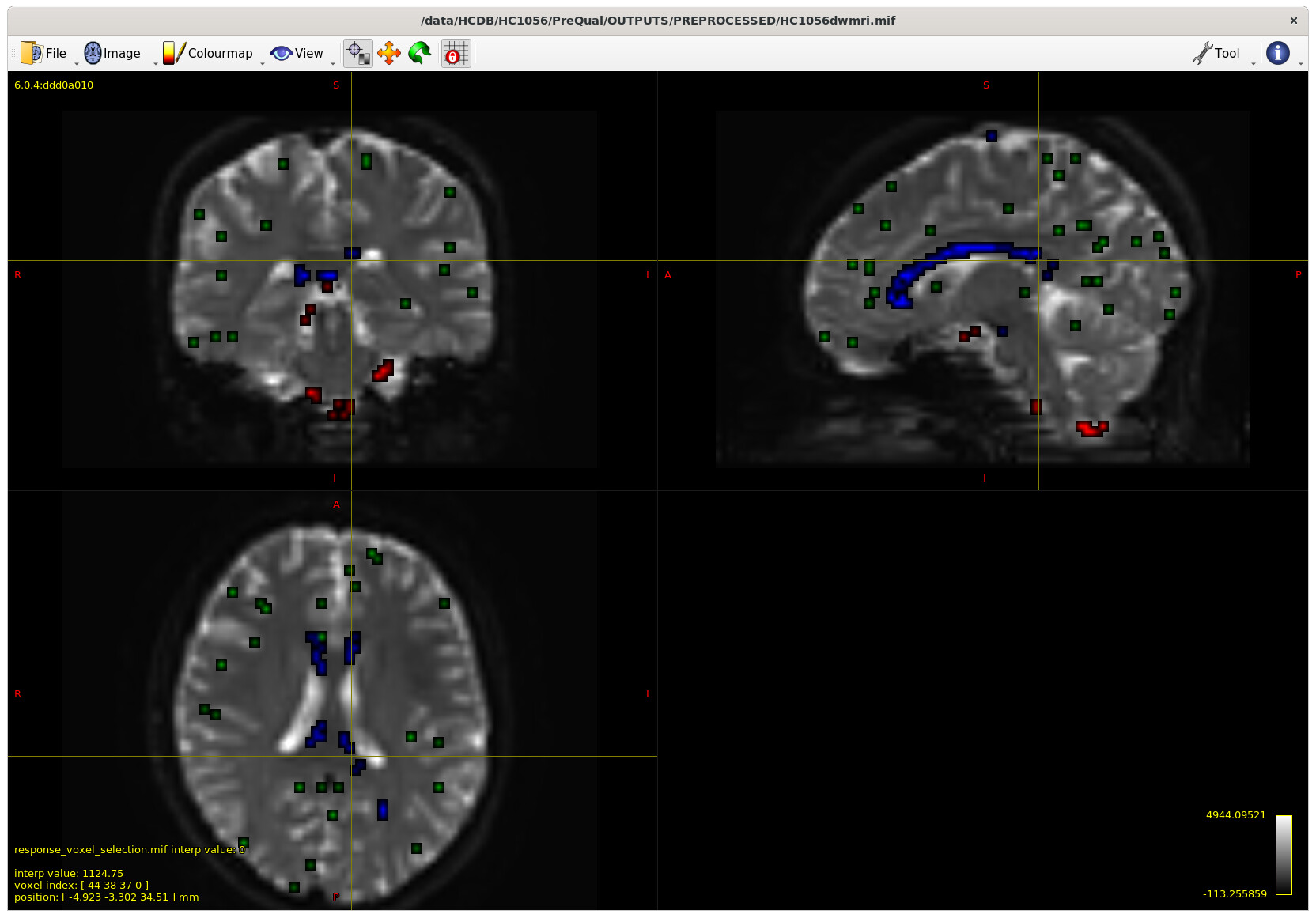
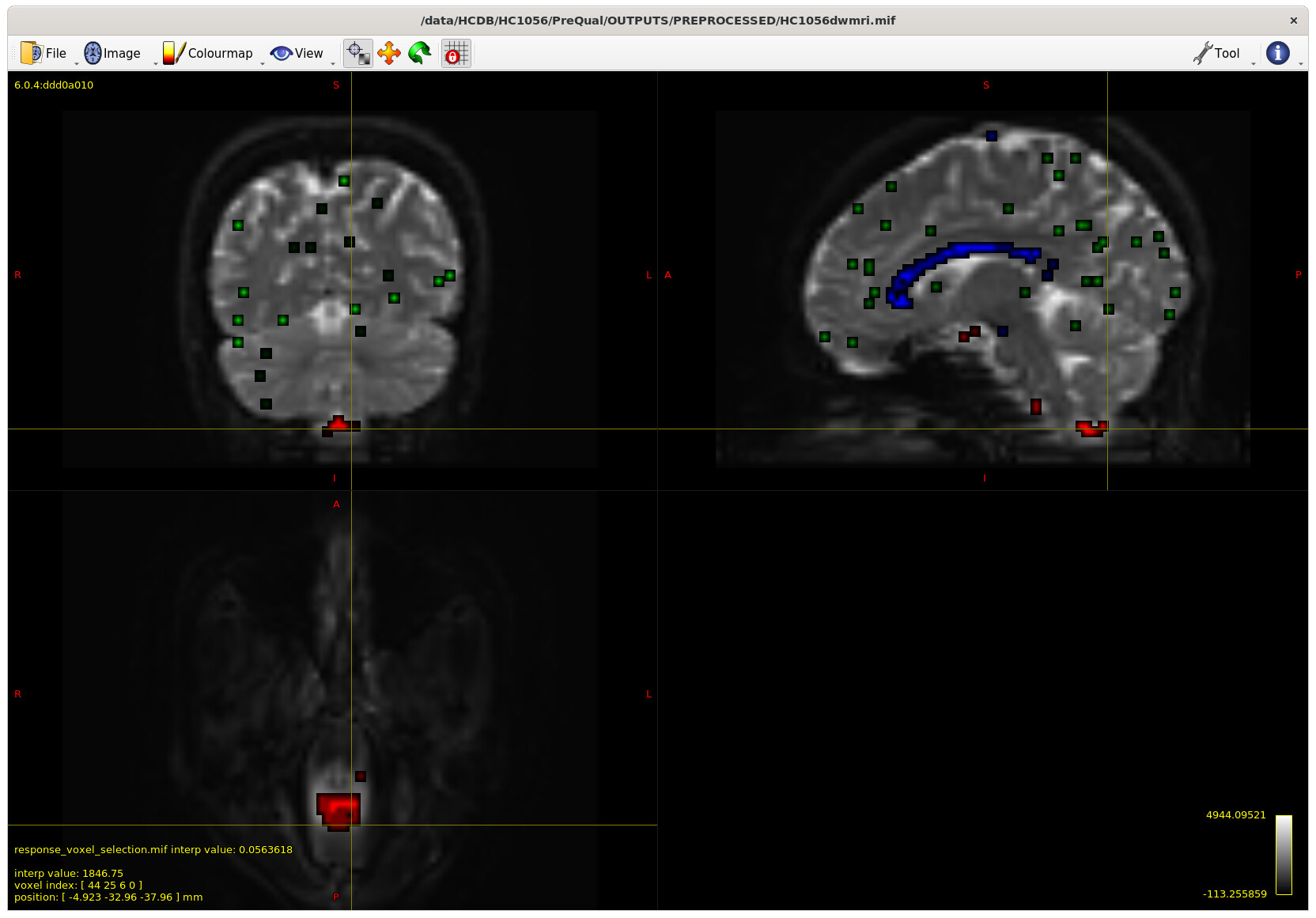
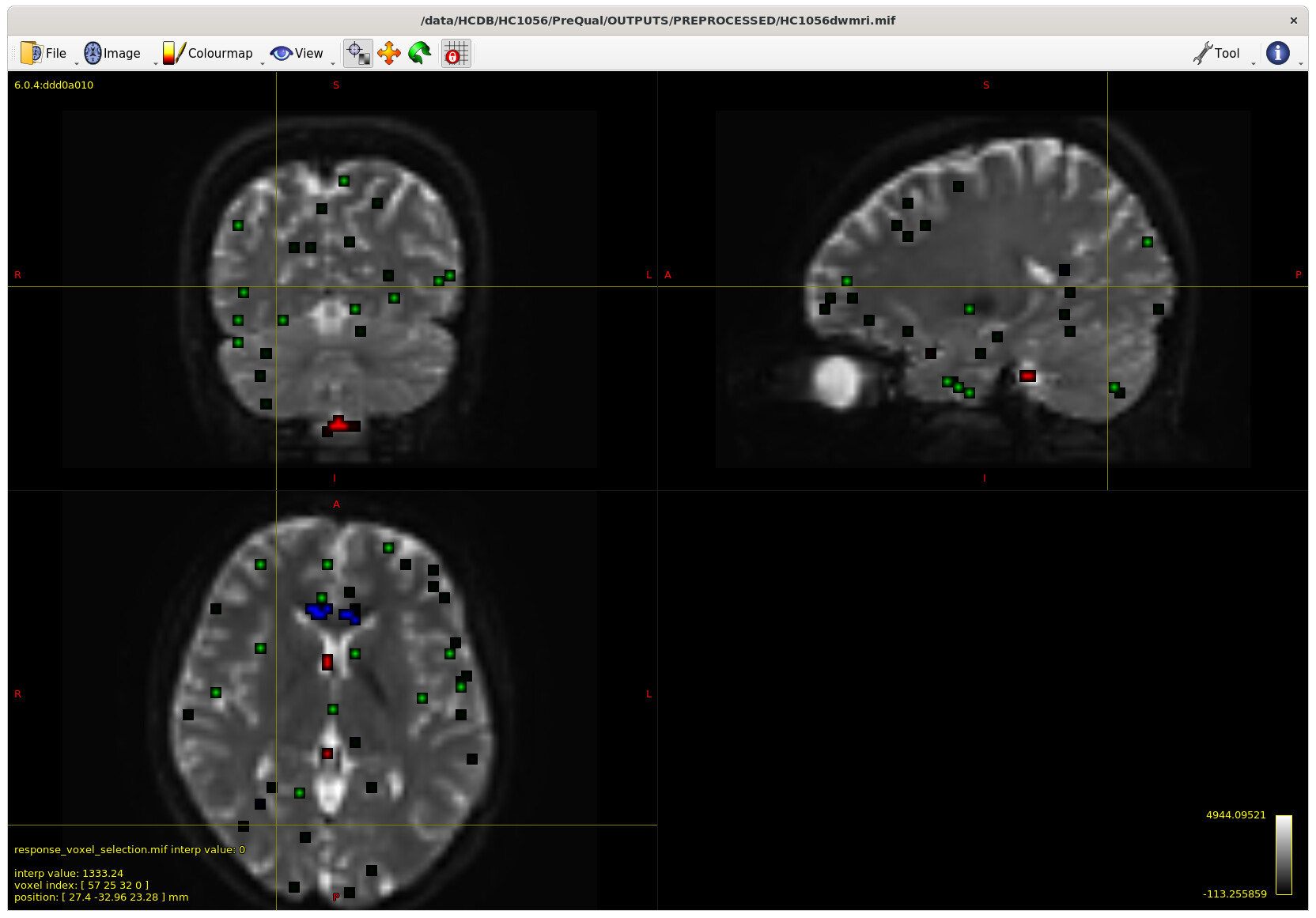
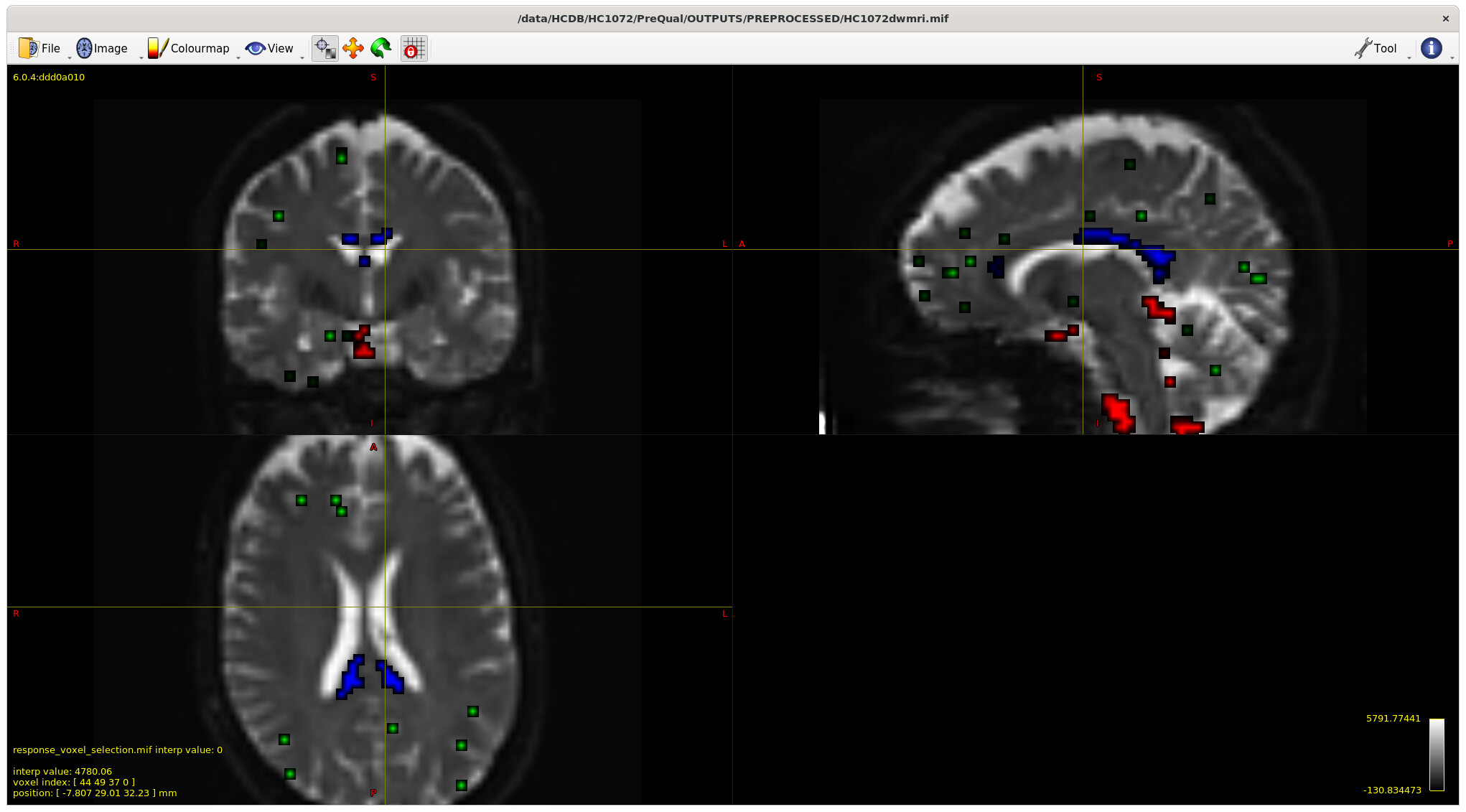
Example with larger ventricles on an older participant:
I moved on to create the average tissue response functions (with 40 subjects), which again look as expected.
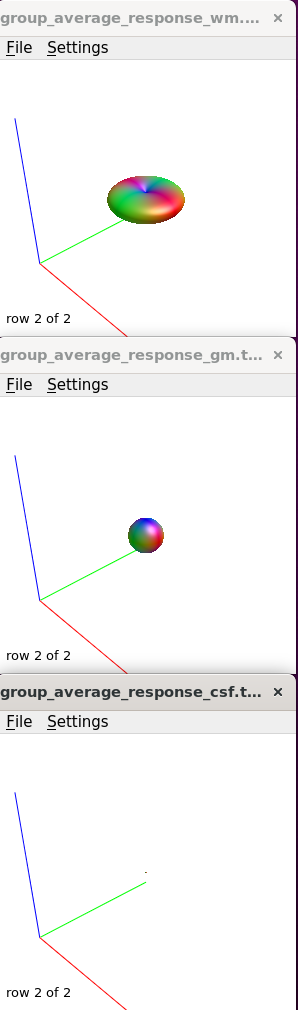
Upsampled to 1.25mm (masks too, and checked these to make sure they were good fits), then used SS3T (https://3tissue.github.io/, all default parameters) with my average response functions to generate the 3T FODs. I checked that the wm FOD’s were in the wm as expected per: MRtrix Tutorial #5: Constrained Spherical Deconvolution — Andy's Brain Book 1.0 documentation
However… it looks like I have some small wm FOD’s appearing in the CSF? Is this normal, or does this suggest an issue with my response functions (perhaps related to above, not sampling a good area for CSF in the response functions?)

Bias Field Correction and Intensity Normalisation
I continued on anyway just to see how the normalization results would turn out. Since the mask fit looked pretty tight to the brain (see above)… I used the upsampled mask for mtnormalize as it was, without erosion. I am wondering if it is important to actually erode the mask just in case for this step even if it’s a good brain fit?
Study specific unbiased FOD template
Had some issues with cropping of my population template – but that’s being resolved here: population template FOV cropped issue
Registration
I registered participants to the resulting population template from the initial fix provided (average_inv = None ) and checked the registrations – they looked good.
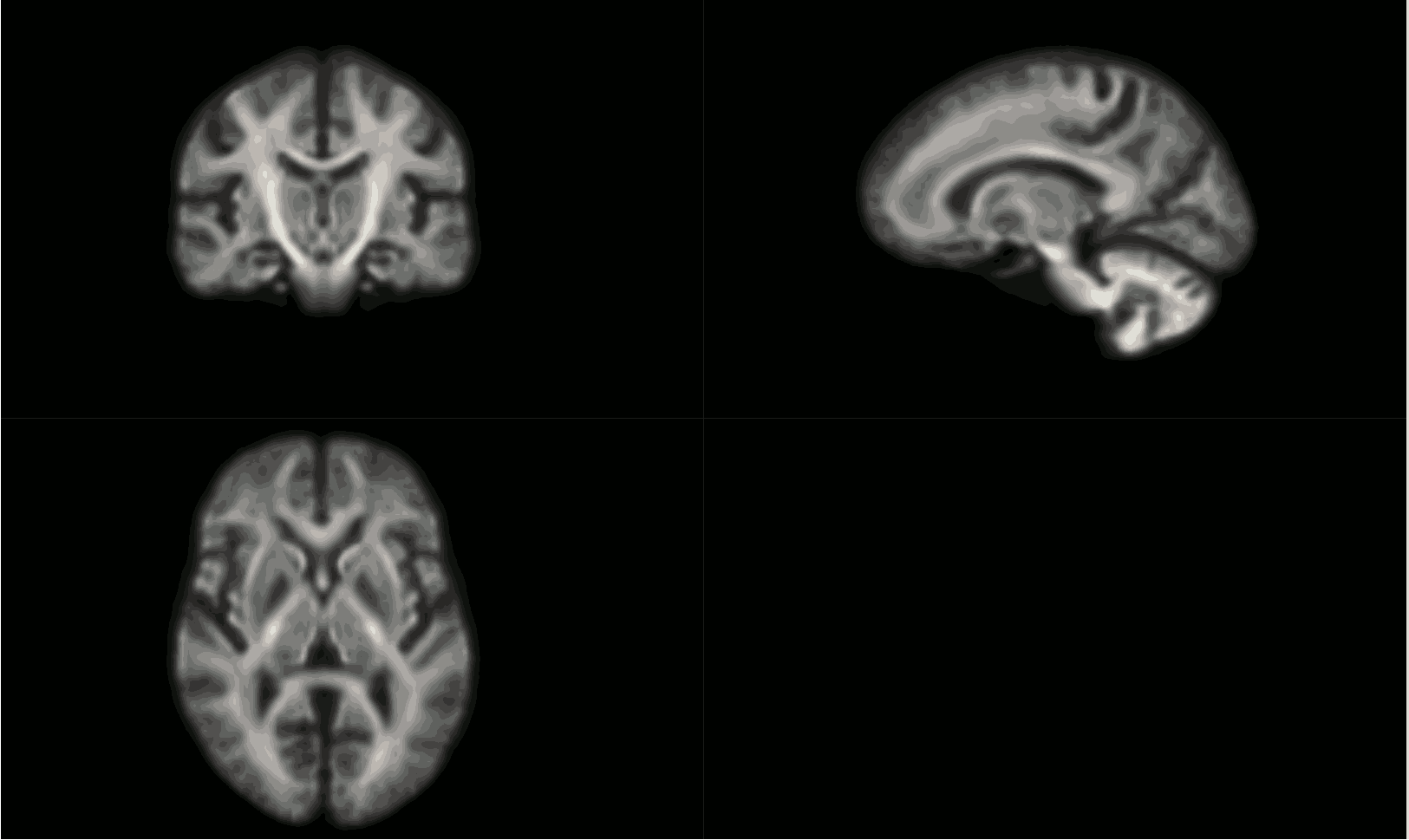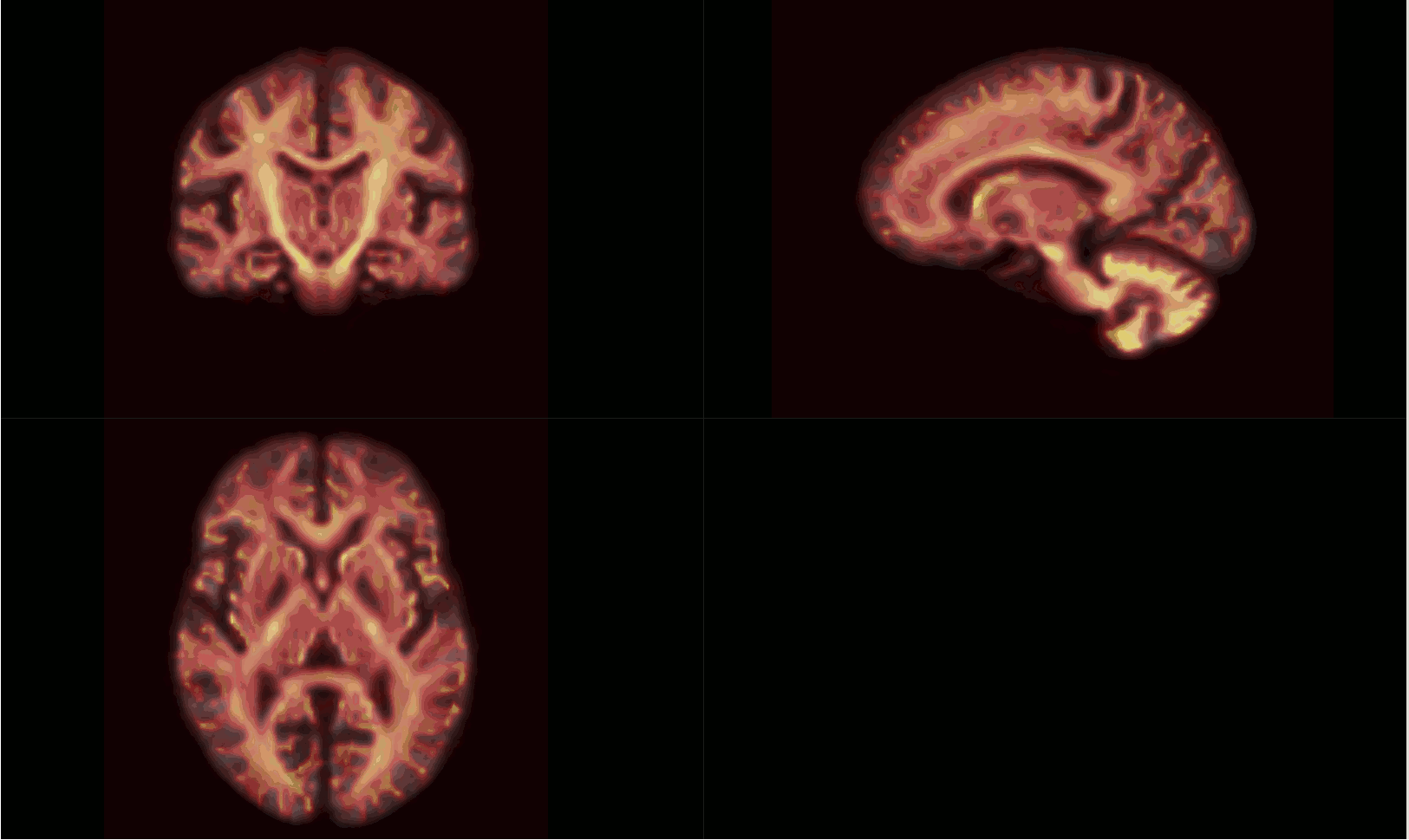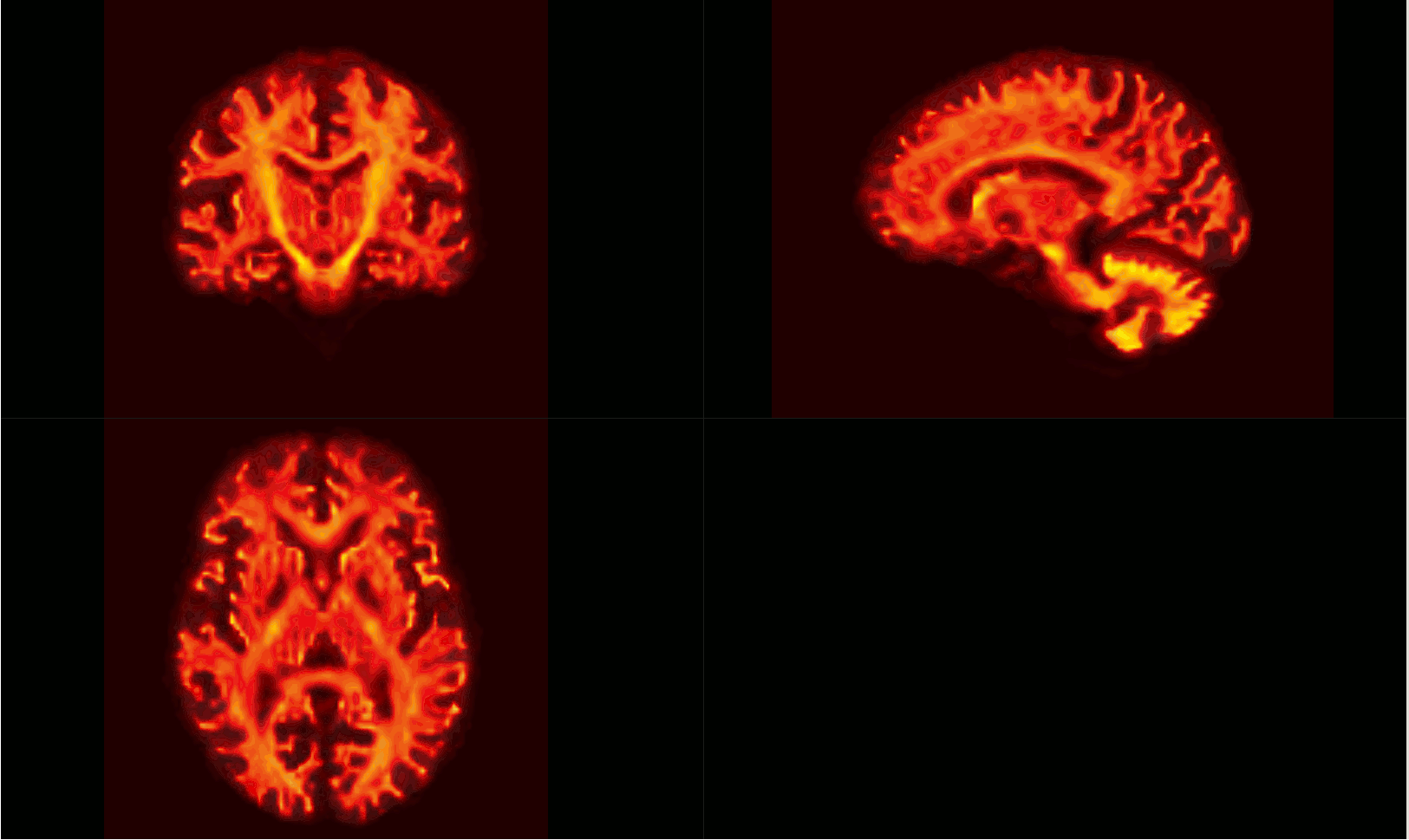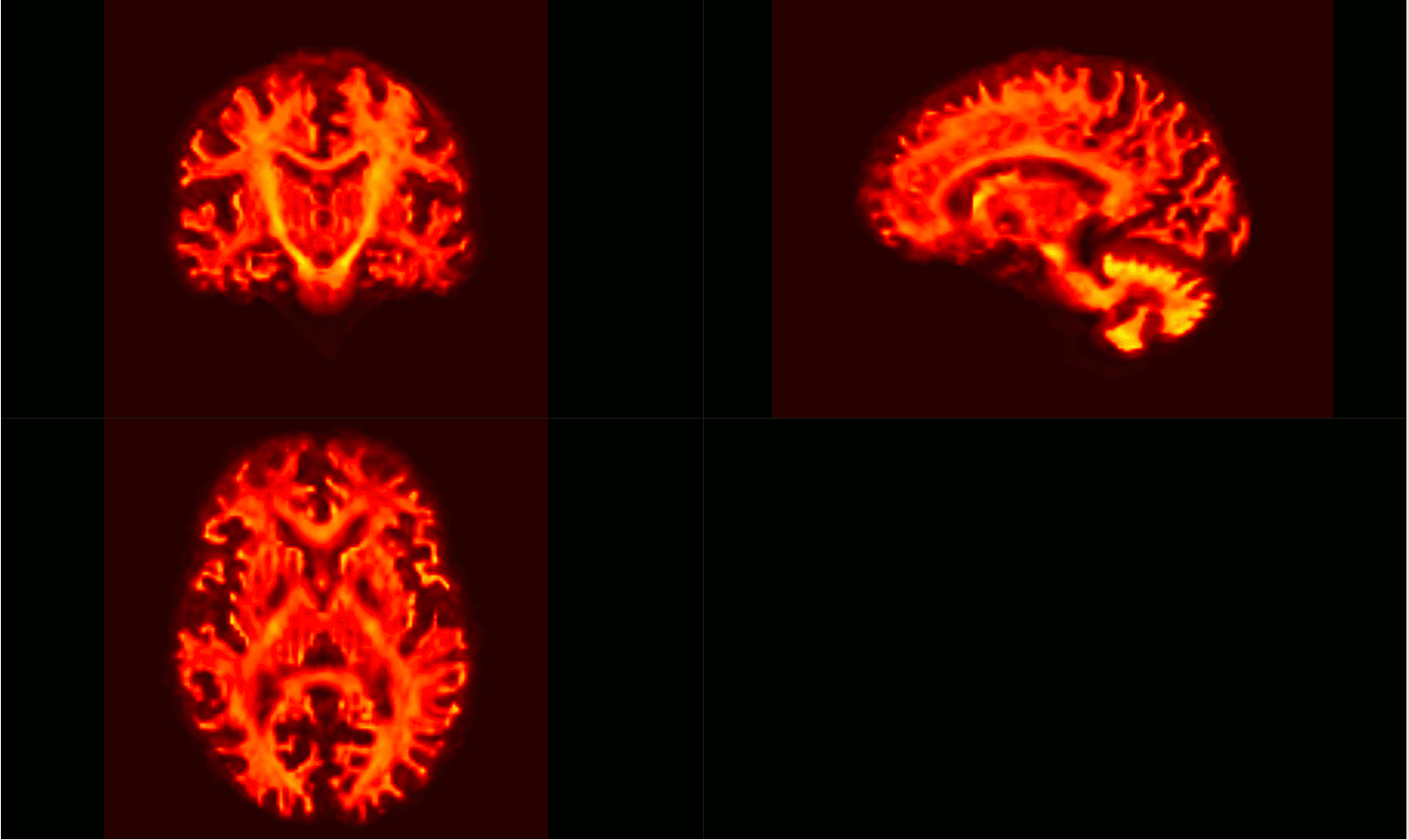
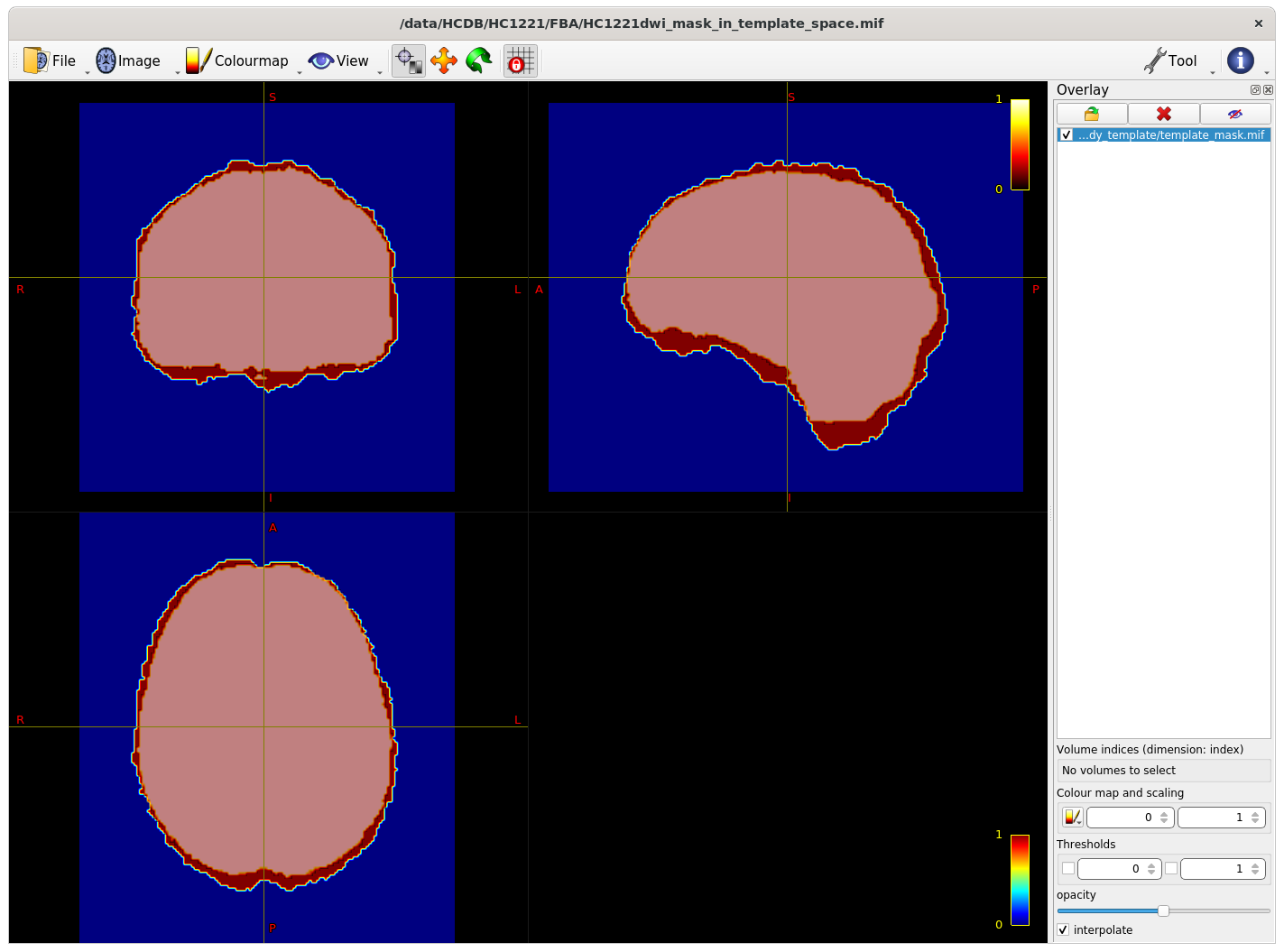
The intersection of all subject masks in template space included all areas of interest for me (example below - light pink is intersection, red is one subject).
Whole brain tractography on the template
Moving past the fixel steps for now – I started generating streamlines for connectivity-based fixel enhancement (CFE) starting with a cutoff of 0.06 as suggested – but many streamlines appeared in the CSF, so I played around with the cutoff (value in bottom right of gif) to see how much I could improve this.
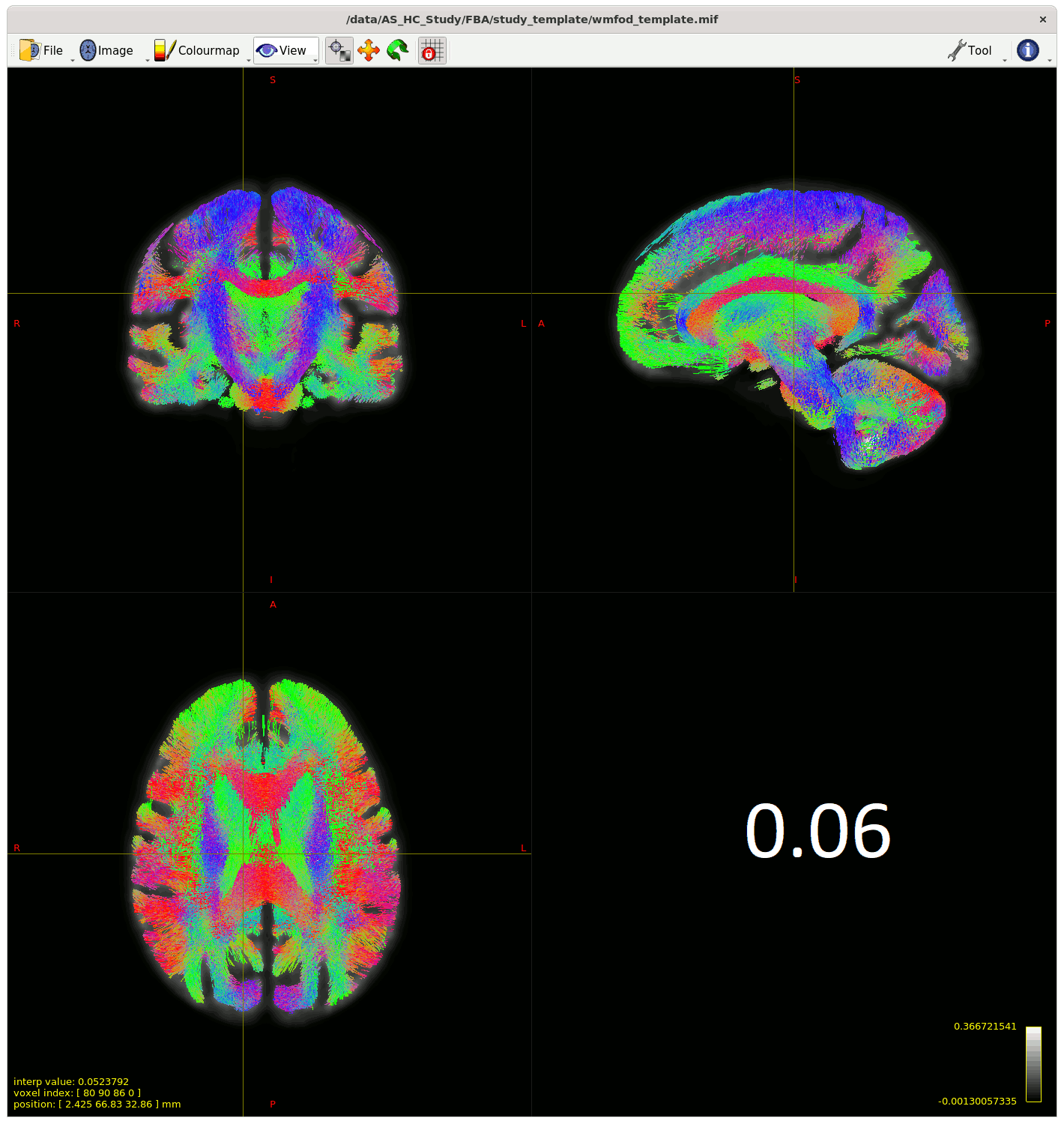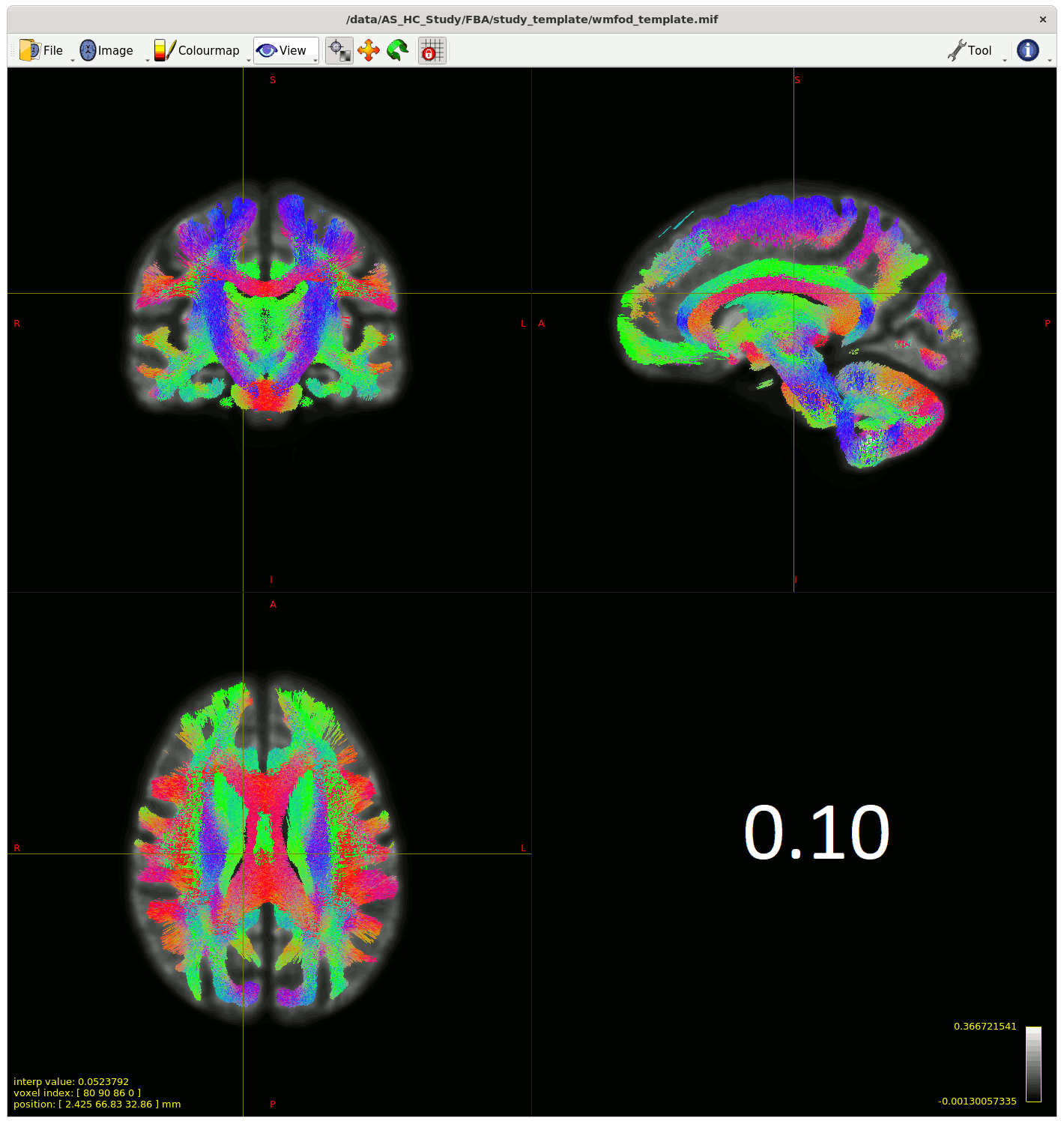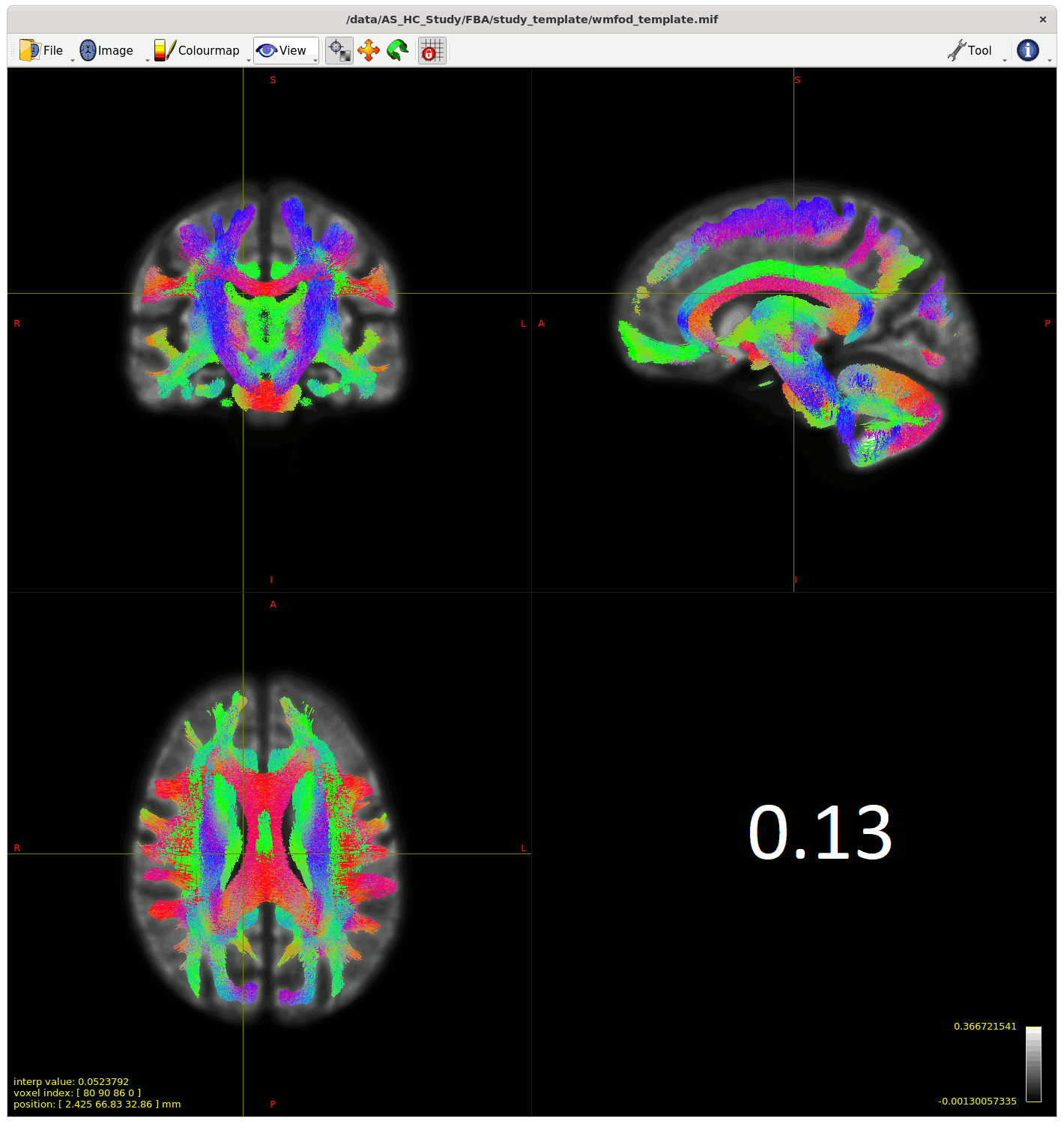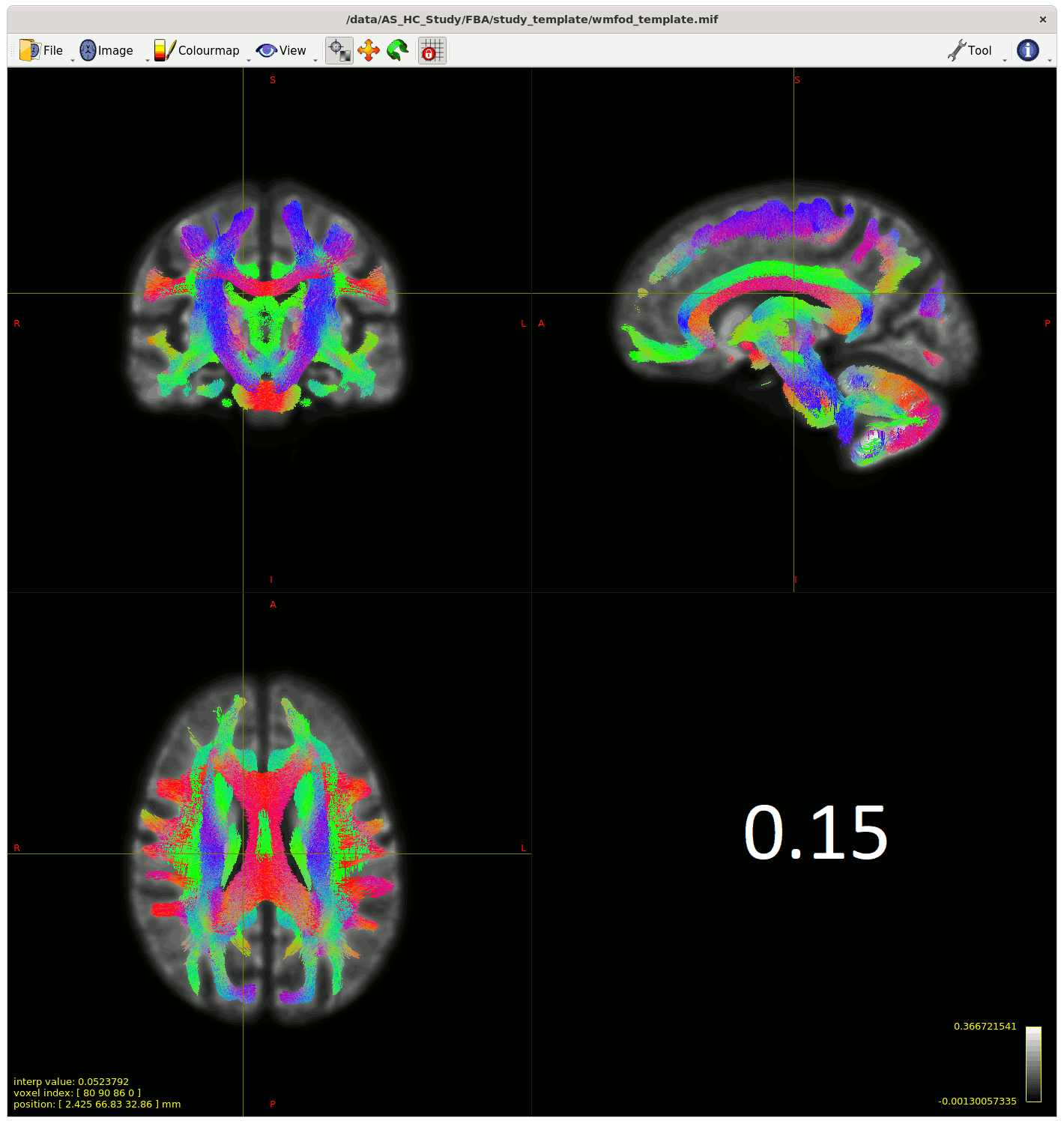
My question is (assuming I can’t improve the streamlines in the CSF from changing something earlier in the pipeline) , does the 0.13 cutoff look decent ? Or is there another way to improve this, somehow using ACT on the template for example?
Thanks a lot for your time and sorry if this was answered already elsewhere